Abstract
Sex development plays a fundamental role in determining the physical attributes of our bodies, the structure of our brains, our behavioral tendencies, and our self-concept. Understanding the development of sex differences has informed and been informed by the advances in the molecular dissection of the pathways responsible for disorders/differences of sex development (DSD). New genes have been identified, allowing for rapid diagnosis, understanding of the pathophysiology, and prediction of future fertility. New genomic technologies, chromosomal microarrays, and next-generation sequencing are revolutionizing the approach to diagnosis of DSD.
In parallel with the progress in the biology of sexual development, the management of individuals with disorders of sexual development has started a gentle revolution. The clinical approach to DSD is slowly entering the era of evidence-based medicine, with emphasis on early genetic diagnosis, involvement of patients and families in a patient-centered decision-making process, and consideration of long-term health-related quality-of-life outcomes.
This chapter (1) reviews the molecular and cellular mechanisms of sex development; (2) presents a classification of DSD, detailing presentations, etiology, diagnostic methods, and recommended management for each condition; (3) includes comprehensive tables detailing syndromic forms of DSD and diagnostic criteria (endocrine, genetic, anatomical), using a modern nomenclature; and (4) offers updated recommendations for management of DSD patients, including considerations about diagnosis, hormonal therapy, gender, fertility, cancer risk, and mental health.
Keywords
Disorders/differences of sex development, DSD, sex, gonad, exome sequencing, testis, ovary, ovotestis
From the moment of our conception, each of us has a sex. Our individual sex has a major role in determining the physical attributes of our bodies, the structure of our brains, our behavioral tendencies, and our self-concept. Understanding the development of sex differences has informed and been informed by the advances in the molecular dissection of genes responsible for disorders of sex development (DSD). New genes have been identified, allowing for rapid diagnosis, understanding of the pathophysiology, and prediction of future fertility.
In parallel with the progress in the biology of sexual development, the management of individuals with disorders of sexual development has started a gentle revolution. From a practice based on the opinions of a few leaders in the field, the clinical approach to DSD is slowly entering the era of evidence-based medicine, under the pressure of patient advocacy groups, who have been highly instrumental in establishing a dialogue between practitioners and their patients. As a result, physicians are beginning to understand the difficulties involved in defining normalcy, as well as the ethical dilemma of acting medically on children before they reach an age of consent.
For the first time, in 2005 a consensus on the management of “intersex” disorders emerged, bringing together experts from a variety of fields (endocrinology, genetics, surgery, psychology, advocacy groups). The consensus statement provides guidelines in all aspects of management of “intersex,” including genital surgery, the requirement of a multidisciplinary team and of mental health professionals, and the diagnostic approach. This chapter reflects the advances made in the field as well as the recommendations of the consensus statement (see also Chapter 17 ). One area that changed considerably was the nomenclature of intersexuality. The term disorders of sex development was proposed, as defined by “congenital conditions in which development of chromosomal, gonadal or anatomical sex is atypical.” This broad definition replaces the word intersex, which has social connotations and reflects a concept of sexual identity. It includes patients who do not necessarily have ambiguous external genitalia (such as complete androgen insensitivity, or Turner syndrome) and yet belong to the broad family of DSD.
The new nomenclature was guided by the following principles. First, although a modern categorization should integrate the important progress in molecular genetic aspects of sex determination and differentiation, it should not overemphasize one particular aspect of the biology of sex (for instance, gonadal sex) and should accommodate the spectrum of phenotypic variations. Second, terms should be as precise as possible and should reflect the genetic etiology when available. Finally, the new nomenclature should be understandable by patients and families and should be psychologically sensitive. In particular, gender labeling in the diagnosis should be avoided, and use of the words hermaphrodite, pseudohermaphrodite, and intersex should be abandoned because they either are confusing or have a negative social connotation that may be perceived as harmful by some patients and parents.
The new nomenclature, which will be used in this chapter, is summarized in Table 16.1 , with an example in Table 16.2 . Another recent development has been the advent of next-generation genomic sequencing. The mapping of all human genes through the Human Genome Project has helped identify the causative genes at several known DSD loci and reclassification of many syndromes after it was uncovered that they shared a common genetic etiology. The availability of exome sequencing in the clinical realm has also ushered in a change in paradigm in DSD diagnosis. With exome sequencing and targeted DSD gene panels available at decreasing cost and with turnaround time similar to that of some endocrine tests (4 weeks for exome at some testing centers, and down to 1 week for urgent cases), prioritizing genetic testing as the first-tier diagnostic tool for the assessment of DSD patients is quickly becoming a reality in clinical practice.
| Current | Previous |
|---|---|
| DSD | Intersex |
| 46,XY DSD | Male pseudohermaphrodite, undervirilization of an XY male, and undermasculinization of an XY male |
| 46,XX DSD | Female pseudohermaphrodite, overvirilization of an XX female, and masculinization of an XX female |
| Ovotesticular DSD | True hermaphrodite |
| 46,XX Testicular DSD | XX male or XX sex reversal |
| 46,XY complete gonadal dysgenesis | XY sex reversal |
| Sex Chromosome DSD | 46,XY DSD | 46,XX DSD |
|---|---|---|
| 45,X (Turner syndrome and variants) | Disorders of gonadal (testicular) development:
| Disorders of gonadal (ovarian) development:
|
| 47,XXY (Klinefelter syndrome and variants) | Disorders of androgen synthesis or action:
| Androgen excess:
|
| 45,X/46,XY (mixed gonadal dysgenesis, ovotesticular DSD) | ||
| 46,XX/46,XY (chimeric, ovotesticular DSD) |
Mechanisms of Human Sex Development
Human sex development occurs in two main steps during embryonic development:
- ◆
Sex determination: Expression of the Y-chromosome gene SRY triggers transformation of the bipotential gonad into a testis in males. The absence of this signal in females results in development into an ovary in females.
- ◆
Sex differentiation: The developing testis secretes hormones that trigger differentiation of wolffian structures, phallus growth, and testicular descent. The absence of testosterone and anti-müllerian hormone (AMH) in females allows for development of müllerian structures and typical female external genitalia.
- ◆
DSD can result from anomalies at any stage of these complex developmental pathways.
Sex determination refers to the developmental decision that directs the orientation of the bipotential, undifferentiated embryo into a sexually dimorphic individual. In mammals this decision occurs during the development of the gonads. In 1947, physiologist Alfred Jost performed a series of elegant experiments showing that all mammalian embryos that are castrated early in development and reimplanted into the uterus develop into females, regardless of their genetic sex. Unilateral grafting of a testis in the embryo results in ipsilateral masculinization of structures, demonstrating that testis-specific factors trigger development of male internal genitalia ( Fig. 16.1 ). This study established the current paradigm of sex determination and differentiation in placental mammals: genetic sex determines gonad development, and gonadal sex in turn governs anatomical sex.
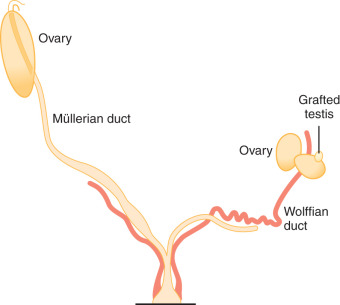
At conception, genetic sex is determined based on whether one inherits an X or a Y chromosome from one’s father. Sex determination is defined by the developmental choice of the bipotential and undifferentiated gonad to become either testis or ovary. Following this sex determination decision, the process of sex differentiation begins and the testes start producing the male hormones testosterone (T) and AMH, which are responsible for male internal and external genitalia development, while the lack of these hormones result in female internal and external genitalia ( Fig. 16.2 ).
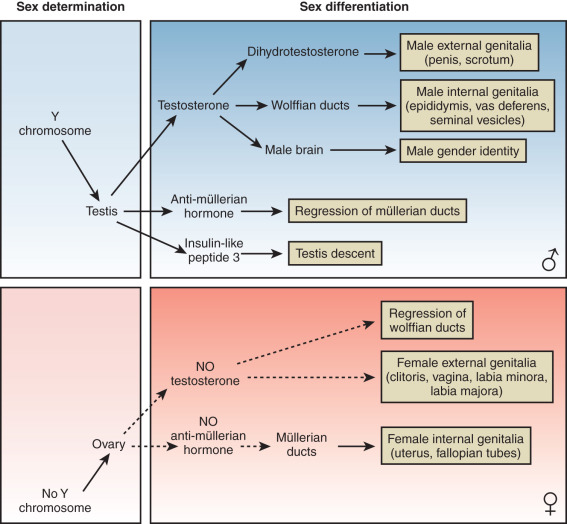
The molecular mechanisms of mammalian sex determination are still incompletely understood. About half of DSD cannot be explained at the molecular level yet, suggesting the existence of a number of unknown sex-determining genes or mechanisms. The next sections outline the known molecular mechanisms affecting sex determination and sex differentiation.
Sex Determination: Testes or Ovary?
This paradigm for sex development established by Jost led to the search for a sex-determining gene that was a testis-determining factor (TDF). When the karyotypes of patients with Klinefelter syndrome (who are male) and Turner syndrome (who are female) were determined to be 47,XXY and 45,X respectively, it became clear that the Y chromosome was sex-determining and that TDF had to be located on the Y chromosome.
In the early 1990s, a series of elegant experiments found SRY to be the elusive mammalian testis-determining gene. Positional cloning located a 35-kb fragment of the Y chromosome translocated to the X chromosome, explaining the presence of male gonadal tissue in 46,XX testicular and ovotesticular DSD patients. Further analysis defined a conserved sequence within this region. Sry gene expression analysis revealed a male-specific increase in transcript consistent with the earliest known divergence of male and female gonadal development. Furthermore, XX mice transgenic for a 14-kb fragment containing Sry developed testes and the full male phenotype. This was followed up with the demonstration that deletions and mutations in SRY / Sry result in XY sex reversal in both humans and mice.
In humans, 8 weeks after conception the bipotential gonad progresses toward testis organogenesis in the presence of SRY and can be recognized on ultrasound by week 10. Factors expressed earlier than SRY prime the bipotential gonadal ridge to differentiate in the male pathway. Mutations in the mitogen-activated protein (MAP)-kinase signaling pathway result in 46,XY DSD with partial or complete gonadal dysgenesis (GD) and is important in promoting SRY expression within the developing gonad. Chromobox-2 (CBX2) has also been shown to be required for Sry expression in mouse sex determination and a mutation was found in a 46,XY female patient.
SRY expression in the Sertoli cell precursors initiates testis determination by activating downstream effectors such as SOX9 ( SRY -related high-mobility group [HMG]-box 9). Sertoli cell proliferation and organization into tubular cords is directed by SOX9 and SF1/NR5A1 (steroidogenic factor-1) . At 9 weeks of gestation, after the organization of discrete tubules, Sertoli cells begin the secretion of AMH. NR0B1/DAX-1 is required in single copy for testes development as point mutations result in hypogonadotropic hypogonadism, whereas two copies in XY fetuses result in XY GD by repressing testis organogenesis. Another gene involved in sex determination is Desert hedgehog (DHH) , which up-regulates NR5A1/SF1 and in turn regulates Leydig cell proliferation.
The cellular fate of murine ovarian and testicular tissues is not sealed at the point of sex determination. In murine models of sex determination, cellular fates of the testis or ovary are actively maintained throughout adult life by DMRT1 (Doublesex and mab-3 related transcription factor 1) and FOXL2 (Forkhead box L2), respectively. This lack of finality in the gonadal cell fate determination indicates that cell fate is carefully cultivated throughout adulthood, and if the factors important in maintaining cell fate are mutated, it may theoretically lead to decreased secondary sex characteristics and infertility.
Relative to testis organogenesis, there is much less information on the genes involved in ovary organogenesis. Studies suggesting that ovary development does not require a large number of genes are incomplete in that they look at time points that are important in testis determination but may not be critical in ovarian determination. Despite this, researchers have uncovered several autosomal genes required for proper ovarian development, including WNT-4 , RSPO-1 (R-spondin-1), and FST (follistatin), and the number of genes linked to ovarian dysgenesis and premature ovarian failure has exploded in the past few years.
Sex Differentiation
The type of internal ( Fig. 16.3 ) and external ( Fig. 16.4 ) genitalia is decided by the presence of a functional testis. Therefore the testis actively governs sex differentiation. Conversely, the growth of female genitalia is the default process in sexual differentiation. The bipotential ductal system consists of the müllerian and wolffian ducts, which give rise to female and male internal genitalia, respectively.
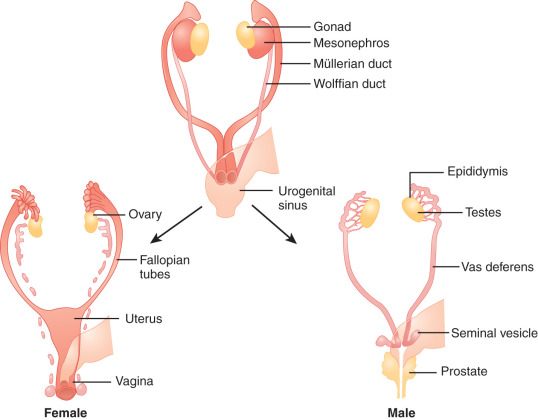
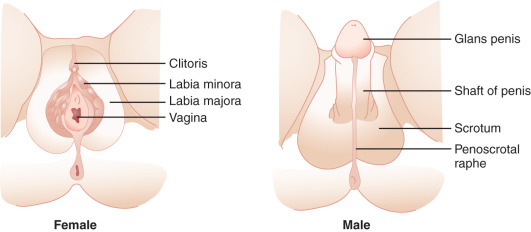
Hormones secreted from testes are essential to the development of male internal and external genitalia. Normally developed testes have both Sertoli cells and testicular cords. Sertoli cells secrete AMH, causing müllerian duct regression. At the same time, Leydig cells, the steroidogenic cells of the testes, secrete testosterone and INSL3 (insulin-like 3), which promote development of wolffian structures (epididymis, vas deferens, and seminal vesicles) and mediate transabdominal descent of the testes to the internal inguinal ring, respectively. Dihydrotestosterone (DHT), a more potent androgen converted from testosterone by the enzyme 5α-reductase, mediates the development of the male external genitalia. Except for phallic growth and inguinoscrotal descent in the third trimester, male sexual differentiation is essentially complete by week 14 of gestation. Defects in labioscrotal fusion and urogenital sinus growth cannot be modified by high doses of androgens after week 14.
In females, the absence of a testicular tissue and its associated hormones, such as AMH or testosterone, allows müllerian structure development (fallopian tubes, uterus, upper vagina) while wolffian ducts regress, around 12 weeks of gestation. Finally, in the absence of DHT, no virilization of external genitalia occurs.
Disorders that can cause ambiguous genitalia but are not associated with dysgenetic gonads, such as androgen insensitivity syndrome (AIS) and congenital adrenal hyperplasia (CAH), are considered disorders of sexual differentiation, not disorders of sex determination.
DSD can present as an isolated urogenital anomaly or as part of a multiorgan syndrome. Gonadal and external genitalia phenotypes associated with syndromic forms of DSD are detailed, with their genetic cause, in Table 16.3 (see Hutson et al. 2014 in the Top References for a classification of syndromic DSD by affected biological process). Overview of the diagnostic differences in DSD, including both disorders of sex determination and disorders of sex differentiation, are presented in Table 16.4 .
| DSD | Biochemical Changes | Differentiating Features | Major Genetic Diagnostic Criteria | |
|---|---|---|---|---|
| 46,XY disorders of gonadal development | 46,XY pure/partial/mixed gonadal dysgenesis | Pure/partial/mixed ↑FSH, LH Nml to ↓ AMH No ↑ with hCG NmL adrenal hormones and precursors Pure ↓↓; T, DHT, E2 ↓↓ AMH Partial/mixed ↓ T, DHT, E2 ↓ T, DHT, E2 | Presence of partial testicular function (T, AMH) points toward partial or mixed GD. However, histologic examination of testes after prophylactic gonadectomy can differentiate between pure, partial, and mixed GD. There is little genotype-phenotype correlation; however, the presence of mosaicism 45,X/46,XY is often associated with mixed GD. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Genetic mutations in SF1, SRY, MAP3K1 provide a definitive genetic diagnosis. If partial/mixed GD suspected, patient should be tested for mosaicism. CGH should be performed to look for copy number variants as a source for XY, GD. |
| Testicular regression | ↑ FSH, LH ↓↓ T, DHT, E2 ↓↓ AMH No ↑ with hCG | Complete absence of gonad or fibrosis of gonad (as opposed to streak or dysgenetic testes in GD patients). Completely virilized male phenotype. | Fibrous nodules on laparoscopic exam (not streak or dysgenetic testes) | |
| Disorders of sex differentiation: Disorders in androgen synthesis and action | StAR deficiency; P450scc deficiency | ↑ Renin ↓ Aldo ↑ K ↓ Na ↓ All adrenal hormones ↓17-OHP | Presence of lipid vacuoles in adrenals on histology. P450scc deficiency does present with enlarged adrenals. No HTN and hyperkalemia differentiates from CYP 17 deficiency | Presence of lipid-filled vacuoles on histology; clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Identification of mutations in the StaR or P450scc genes gives a definitive diagnosis. |
| 3βHSD type II | ↑ Renin ↓ Aldo, F ↑ K ↓ Na ↑ Ratio Δ -17-pregnenolone: cortisol ↓ Δ4A, T | Baseline and ACTH-stimulated ratios of Δ -17-pregnenolone:cortisol consistently distinguished between affected and nonaffected patients. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Mutations, deletions, and insertions in the 3βHSD type II gene provide a definitive genetic diagnosis. | |
| 17α-hydroxylase/17,20-lyase | ↓ Renin ↓ Aldo, F ↓17-OH Progesterone ↑ LH, FSH ↑ Progesterone, DOC, B ↑ Na ↓ K ↓ DHEA-S, Δ4A, T; no response to hCG stim | Hypertension and hypokalemic alkalosis in the presence of low 17-OH progesterone. Isolated 17,20-lyase deficiency would have decreased levels of sex steroids with normal mineralocorticoids and normal glucocorticoids. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Identification of mutations of CYP17 or CYPB5 gene provides a definitive diagnosis. | |
| P450 oxido-reductase | ↑ 17OH-progesterone ↑ Progesterone ↓ F, DHEA-S ↓ Δ4A, T | Hypertension, in the presence of elevated 17-OH progesterone (differentiates from CYP17 deficiency); sometimes presence of Antley-Bixler skeletal malformations. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Mutations in CYP450 oxidoreductase provide a definitive diagnosis. | |
| 17βHSD Type 3 | Nml to ↑ Δ4A ↑ ratio Δ4A/T (>15) ↓ T, DHT | Differentiated from 5α-reductase type 2 by levels and ratios of serum hormones. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Sequencing of the 17βHSD gene for deletions, insertions, and point mutations. | |
| Leydig cell hypoplasia | ↑ LH Nml FSH ↑ AMH ↓ T, DHT, E 2 ↓ hCG response Nml Δ4A/T ratio | To differentiate LCH from GD, AMH is used as a marker of testicular function. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Deletions, insertions, and point mutations in the LHR gene provide a definitive diagnosis. | |
| 5α-reductase type 2 mutations in the backdoor pathway to androgen activity | Nml FSH, LH Nml T, E 2 ↓ DHT ↑ ratio T/DHT (>30) Nml ratio T/DHT (>30) Nml AMH Nml cortisol ↓ DHEA-S after hCG ↑ 17OH-pregnenolone | Development of male secondary sex characteristics in puberty with fine and sparse facial hair. These can be differentiated clinically from 17βHSD and AIS by the lack of gynecomastia during puberty. Similar in phenotype to 17βHSD and 5-alpha reductase deficiency but can be differentiated by serum hormone levels. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Point mutations, deletions, insertions, and parental isodisomy in SRD5A2 gene provide a definitive genetic diagnosis. Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Mutations in AKR1C2 and AKR1C4 gene that result in a reduction of their enzymatic activity provide a definitive genetic diagnosis. | |
| Androgen receptor | Nml FSH, LH (PAIS) ↓ FSH, LH (CAIS) Nml to ↑ AMH Nml Δ4A, T, DHT; nml ratios ↑ ↑ hCG response | Female phenotype with breast development at puberty, with sparse pubic and axilla hair | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Sequencing of AR gene looking for single amino acid substitutions, which account for 90% of reported cases. | |
| AMH/AMHR | Nml hormonal profile | Presence of both müllerian and wolffian derivatives, usually discovered incidentally. | ||
| 46,XX disorders of sex determination | XX Testicular/ovo-testicular DSD | ↑ FSH, LH ↓ T, DHT Nml AMH No ↑ with hCG | Only way to differentiate between 46,XX testicular and ovotesticular DSD is by complete gonadal histologic examination looking for the presence of ovarian tissue. | Testicular DSD: FISH for presence of SRY or SOX9 or if clinical phenotype appropriate, sequence RSPO1 gene. If no molecular diagnostic is successful, CGH should be performed. Ovotesticular DSD: Search for XX/XY mosaicism should be done (30%–33% ovotesticular DSD) and sequencing for SRY should be done (7%–10% of ovotesticular DSD). |
| XX Gonadal dysgenesis | ↑ FSH, LH No ↑ with hCG No AMH | Full female phenotype with amenorrhea and lack of secondary sex development | For cases of isolated 46,XX GD, sequencing for FSH mutations can be performed. If clinical presentation matches, sequence FOXL2 mutation. CGH can also be done to identify delirious duplications or mutations. | |
| Disorders of androgen excess | 21α-hydroxylase | ↑ Renin ↓Aldo, DOC, F↑ K ↓ Na↑ 17OH-progesterone↑ DHEA-S, Δ4A, T | 17-OH progesterone elevated in the absence of hypertension, which is typical in 11βHSD1 deficiency. | Sequencing of CYP21 gene gives definitive diagnosis. Knowledge of the mutation can inform parents about genetic counseling and fetal diagnosis to prevent adverse effects of hyperandrogenism in utero. |
| 11βHSD1 | ↓ Aldo, F, Renin↑ DOC↑ K ↓ Na↑ 17OH-progesterone↑ DHEA-S, Δ4A, T | Differentiate from CYP21 deficiency by presences of hypertension and hypokalemic alkalosis. | Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Mutations in 11βHSD1 gives a definitive diagnosis. | |
| P450 aromatase | ↑ 16OH-Δ4A (maternal)↑ FSH, LH, Δ4A, T↓Estrone, E 2 | The presence of maternal virilization during pregnancy and XX virilization which stops after delivery. | The presence of maternal virilization during pregnancy and XX virilization, which stops after delivery. Clinical whole exome sequencing or Sanger sequencing can be performed to assess for mutations in known DSD genes. Mutations in p450 aromatase gives definitive diagnosis. |
Sex Chromosome Disorders of Sex Development
- ◆
Sex chromosome aneuploidies, in mosaic and nonmosaic forms, result in recognizable syndromes such as Turner syndrome (45,X and associated karyotypes) and Klinefelter syndrome (47,XXY).
Sex chromosome DSD are defined by aneuploidy of the sex chromosomes, X and Y. In disorders of aneuploidy (e.g., Trisomy 21), maternal errors in meiotic nondisjunction during meiosis-I account for the majority of cases. However, in Klinefelter syndrome (47,XXY), maternal and paternal errors are equally likely, and advanced maternal age is not a risk factor for Turner syndrome.
Turner Syndrome
In 1938, a group of females with short stature, primary amenorrhea, and a lack of secondary sexual characteristics was described by Turner. Turner syndrome is classified as a sex chromosome DSD, with the majority having a 45,X karyotype, and the remaining having a 46,XX karyotype with a deletion in part of one X chromosome or having various mosaics involving 45,X and 46,XX, and 46,XY cells or more complex combinations. In 100% of patients with 45,X and 80% of mosaic patients, the universal feature is short stature ( Fig. 16.5 ), with average height less than 148 cm. The genetic locus for short stature in both Turner syndrome and idiopathic short stature lies in the pseudoautosomal region (PAR1) of the X chromosome. This region is deleted in individuals with short stature caused by the lack of one copy of the short stature homeobox gene (SHOX) .
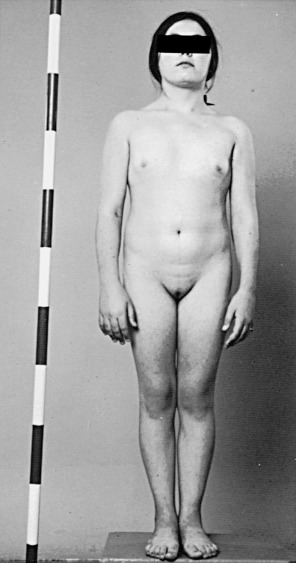
The classic Turner 45,X karyotype is thought to be one of the most common human chromosomal abnormalities and is estimated to occur in 0.8% of all zygotes. However, less than 3% of these zygotes survive to term, and this karyotype is commonly found in spontaneous abortions. The incidence of 45,X karyotypes is approximately 1 in 2500 live newborn phenotypic females.
The gonads of Turner syndrome females do not develop normally, having reduced growth and follicle formation in utero. A normal number of eggs develop in girls with Turner syndrome, but, for unknown reasons, they disappear prematurely. The gonads appear as streaks of white tissue next to the fallopian tubes and are called streak gonads, which on histology have primitive connective tissue stroma without primary follicles. Patients with Turner syndrome have fewer follicles and therefore less estrogen secretion from granulosa cells than normal females, resulting in delayed puberty.
Presentation and Diagnosis of Turner Syndrome
In the prenatal period, fetuses with 45,X DSD and its variants frequently have intrauterine growth restriction. Suspicion for Turner syndrome should be raised if the presence of large septate cystic hygromas, nuchal thickening, short femur, total body lymphangiectasia, or cardiac defects is detected by ultrasound. At birth, Turner syndrome babies often have low birth weights; 30% of babies will present with lymphedema of the upper and lower extremities ( Fig. 16.6 ), an extension of the lymphangiectasia in utero, which disappears in the first few months of life. Additionally, the presence of a webbed neck (pterygium colli) (see Fig. 16.5 ) and dysmorphic features including a high-arched palate, low-set prominent ears, low posterior hairline, epicanthal folds, micrognathia, hypoplastic nail beds, or hypoplastic fourth and fifth metacarpals at birth should raise the suspicion for 45,X DSD. Turner syndrome patients may have increased carrying angle of the arms (cubitus valgus), shield-like chest with wide-set nipples, hearing loss, and a higher frequency of cardiovascular disease such as coarctation of the aorta. Furthermore, the incidence of renal anomalies is between 30% and 50%, the most common finding being a horseshoe kidney, followed by abnormal vasculature.
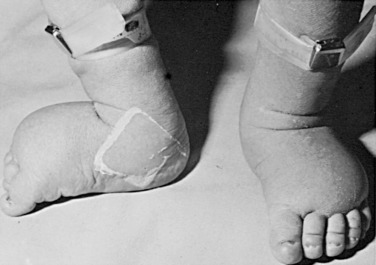
During adolescence, the most common presentation is short stature, amenorrhea, and lack of secondary sexual characteristics. Patients may also have any of the other characteristics mentioned above. 45,X patients have normal intelligence. Depending on the degree of GD, up to 30% of Turner patients undergo some degree of spontaneous puberty.
Diagnosis is made by karyotype, which will show in a 45,X or mosaic variant of cells. If the karyotype appears normal but the presentation is suggestive of Turner syndrome, a fluorescent in situ hybridization (FISH) or chromosomal microarray study should be done because there may be a cryptic deletion in the pseudoautosomal region in one of the X chromosomes. Typically patients with 45,X or 46,XX with a chromosomal abnormality in one X chromosome have more prominent phenotypic features than those who are a mosaic.
Management of Turner Syndrome
For most patients with 45,X DSD, the greatest concern is for short stature and secondary sexual development. The treatment of both of these should be carefully titrated. For cases diagnosed in childhood, the use of low-dose anabolic steroid coupled with growth hormone has been shown to increase final adult height with increased beneficial effects compared to those who started therapy late. Optimal duration and timing of the steroid treatment is still under investigation. At the onset of the age of puberty, high-dose estrogen and progesterone should be given to promote the development of secondary sex characteristics. Estrogens and progesterone should be taken throughout life to prevent complications, such as osteoporosis.
In young patients diagnosed with Turner syndrome, spontaneous puberty is a good indicator of the presence of functional ovarian tissue. Overall, it is estimated that 2% to 5% of patients with Turner syndrome have the potential for spontaneous pregnancy.
A frequent question from families concerns fertility. Despite progress in egg preservation techniques, which could provide hope for fertility in women with Turner syndrome, pregnancies (conceived with either autologous or donated oocytes) carry a high risk of life-threatening cardiovascular complications (aortic dissection, severe hypertension). A complete cardiac assessment is recommended prior to consideration of pregnancy, as well as close monitoring during pregnancy and in the postpartum period.
Klinefelter Syndrome
Klinefelter syndrome is typically associated with hyper gonadotropic hypogonadism and infertility. In utero, the XXY fetal testis has the normal complement of primordial germ cells. However, these germ cells degenerate during childhood, possibly due to a defect in Sertoli cells and germ cell communication during testes maturation. In theory, the etiology of the nondisjunction in Klinefelter can be from maternal meiosis I or II or paternal meiosis II, and thus each situation should contribute to 33% of cases. However, nearly 50% of Klinefelter patients show a paternal origin, with some studies showing increased frequency of diploid XY sperm with advanced paternal age.
Presentation and Diagnosis of Klinefelter Syndrome
Klinefelter syndrome is largely undiagnosed in the general population. In early life, Klinefelter may be diagnosed in boys with behavioral disorders, abnormally small testes, and long legs ( Fig. 16.7 ). The presence of only long lower extremities distinguishes Klinefelter syndrome from the other forms of eunuchoidism that results in equally long upper and lower extremities. In patients with Klinefelter syndrome, IQ typically falls in the normal range; however, it tends to be below that of other siblings. Most patients present in adolescence with small firm testes and hypogonadism with varying degrees of androgen deficiency. In later life, many males present at infertility centers with azoospermia.
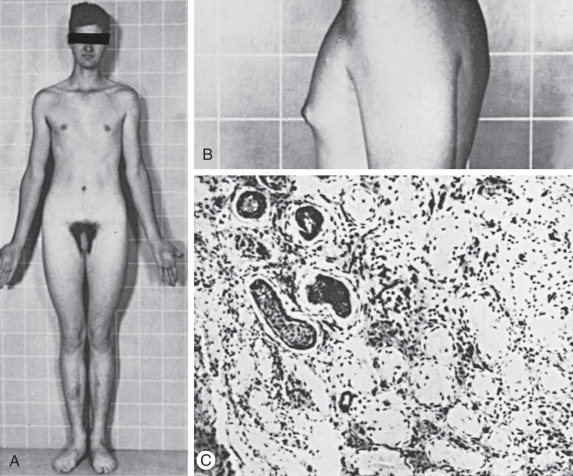
Diagnosis of Klinefelter syndrome is performed by lymphocyte karyotype or chromosomal microarray. Some mosaic cases will only be detected by karyotype of skin fibroblasts and occasionally of testicular biopsy specimens.
Management of Klinefelter Syndrome
Early detection of Klinefelter allows for early intervention for cognitive and behavioral disorders. Current studies are underway exploring the role of androgen replacement therapy in childhood, to help with cognitive and behavioral disabilities. Replacement of androgens also allows for development of masculine secondary sex characteristics, improved self-esteem, and increased libido, strength, and bone mineral density. With regard to fertility, testicular mosaicism is an important factor in determining spermatogenesis and the potential for fertility. The vast majority of 47,XXY patients are azoospermic but, since the advent of surgical testicular sperm extraction and intracytoplasmic sperm injection (ICSI) technology, have the potential to be fertile. It should be noted that there is a higher rate of sex chromosomal hyperploidy and autosomal aneupoloidy in the sperm of Klinefelter patients. However, the risk of passing sex chromosome aneuploidy to the offspring remains unclear. Regardless, preimplantation genetic diagnosis (PGD) or fetal karyotype can identify chromosomal abnormalities in the progeny. Using testicular sperm extraction technology, sperm can be successfully extracted from the testes and injected into a donor oocyte, with a reported fertility success of up to 50%, a dramatic change from the near complete historical infertility of Klinefelter patients.
46,XY Disorders of Sex Development
- ◆
46,XY GD results from abnormal testis development in utero. The absence of secreted hormones results in a spectrum of genital phenotypes from typical female presenting in adolescence with primary amenorrhea to ambiguous external genitalia. Known causative genes include SRY, SOX9, SF1, and MAP3K1. Patients need to be monitored for risk of gonadoblastoma.
- ◆
46,XY disorders of androgen biosynthesis are disorders of sex differentiation that include steroidogenic acute regulatory protein (StAR), 3βHSD, CYP17A1, P450 oxidoreductase (POR), 17β-HSD, and steroid 5α-reductase (SRD5A2) deficiencies. Although all of these are often difficult to distinguish clinically, an early genetic diagnosis is critical because they have different natural histories requiring different management.
- ◆
46,XY disorders of androgen action are mutations in the androgen receptor (AR) resulting in complete and partial AIS (PAIS).
46,XY Gonadal Dysgenesis
Etiology and Pathophysiology of Pure/Complete Gonadal Dysgenesis
XY GD is a result of abnormal testis development in utero. There are three types of GD, pure (or complete), partial, or mixed, all of which can be differentiated by the extent of normal testicular tissue within the gonad and karyotype of the individual. In pure/complete GD, individuals have intraabdominal, bilateral, fibrous streaks that do not secrete AMH or testosterone. Phenotypically, pure XY GD individuals are unambiguously phenotypic females (previously known as Swyer syndrome) but usually internally possess hypoplastic müllerian structures.
Given the primary importance of SRY in human testes determination, it is surprising that mutations in SRY only account for approximately 15% of all 46,XY DSD with pure GD. This suggests the existence of many other genes involved in primary human sex determination. SRY is a transcription factor, but its mechanism of action remains incompletely understood. For instance, it is still unclear whether it acts as an activator or repressor, and its only identified and confirmed target is SOX9, another transcription factor from the Sox (Sry-box) family of genes. This interaction is mediated by the testis-specific Sox9 enhancer, TESCO, and by TESCO-independent mechanisms. To date, there are more than 50 verified mutations within the SRY gene. Mutations that result in streak gonads primarily occur within the HMG box, and cause reduced DNA binding, alterations in DNA bending, or prevents nuclear import of the SRY protein. Larger cytogenetic deletions of Yp that include SRY have also been implicated in XY GD.
Steroidogenic factor 1 (SF1, encoded by the gene NR5A1 ) is an orphan nuclear receptor necessary for adrenal and testicular development. Interactions between NR5A1/SF1 and the transcription factors GATA-binding protein 4 (GATA4) and friend of GATA 2 (FOG2) appear to be necessary for SRY expression in developing testes. Recent studies have underlined the importance of SF1 in testicular development, and nearly 15% of isolated XY GD can be attributed to SF1 haploinsufficiency. However, SF1 mutations with XY GD and adrenal hypoplasia congenita have been reported, which reflect the role of SF1 also in adrenal development. Together, mutations or deletions of SRY and SF1 account for 30% of XY GD, indicating that other genes critical to the testes determination cascade still remain to be discovered.
Exome sequencing has now shown that MAP3K1 variants are frequent in 46,XY DSD patients and may be an underestimated cause. Mutations in genes involved in testis sex determination (SOX9, WT-1, DHH, DMRT1) and duplications of putative “anti-testis” genes (WNT-4, DAX1) are responsible for a minority of all XY GD. Many are also associated with other syndromic features, which are outlined in Table 16.3 .
Etiology and Pathophysiology of Partial or Mixed Gonadal Dysgenesis
Like many developmental disorders, there is a wide range in phenotypic variability in patient presentation. When testes dysgenesis does not involve the entirety of both gonads and the external genital phenotype is ambiguous rather than typical female, the condition is called partial GD . The internal genitalia display varying degrees of wolffian and müllerian development, which correspond with the proportion of the gonads that is dysgenetic or “streak.” The medical literature is often confusing in regard to the difference between “partial” and “mixed” GD, which are often used interchangeably. Partial XY GD refers to intermediate stages of dysgenetic testes, between streak gonads and normal testes, and usually has a 46,XY, nonmosaic karyotype. Mixed XY GD typically refers to a situation in which one gonad is a streak gonad while the opposite gonad is partially dysgenic or a normal testis. Mosaicism for 45,X/46,XY is the most frequent cause of mixed GD, although a minority have a 46,Xi (Yq) karyotype. The evidence of the variable phenotypic spectrum of individuals with 45,X/46,XY mosaicism was reported in a series of 10 45,X/46,XY patients in which four individuals were undervirilized males with bilateral testis, three were diagnosed with mixed GD and genital ambiguity, and three were diagnosed with Turner syndrome. Although not proven, there is preliminary evidence that the percentage of normal testicular tissue and phenotypic maleness are correlated with an increase in the proportion of gonadal Y chromosome.
Familial cases of pure and partial 46,XY GD have been reported. In some of these cases, SRY mutations present with phenotypic variability in which the father and male relatives are phenotypically normal and fertile, while their XY offspring have GD. From these cases, it is clear that autosomal genetic modifiers influence the sex determination cascade and ultimate phenotypic appearance of the individual. There are also cases of fathers who are mosaic for mutant and normal SRY transmitting the mutation to their XY female daughters. Presumably, the normal father’s mosaicism reflects a postzygotic mutation event. These unique cases provide more hints about the molecular basis of sex determination. This is particularly relevant because the genetic cause of about half of 46,XY GD remains unknown.
Presentation and Diagnosis of Gonadal Dysgenesis
Pure XY GD presents as a phenotypic female with normal or tall stature, bilateral streak gonads, delayed puberty, amenorrhea, small or normal müllerian structures, and without signs of Turner syndrome. If patients are not diagnosed at birth (e.g., when in utero karyotype does not match the phenotypic sex at birth) most patients will be diagnosed during adolescence due to pubertal delay and primary amenorrhea. Patients can rarely present with an abdominal or pelvic mass, which is often a gonadoblastoma. Occasionally, XY GD can be part of a constellation of other symptoms, outlined in Table 16.3 .
Individuals with partial or mixed 46,XY GD typically present at birth with varying degrees of masculinization of external and internal genitalia. Depending on the percent of testicular tissue, dysgenetic testes can be found anywhere along the line of testes descent, from the abdomen, and in cases of normal testes, in the scrotum. However, the streak gonad in mixed 46,XY GD is always abdominally located. Patients with partial 46,XY GD usually have female external genitalia with some degree of virilization, such as clitoromegaly or a bifid scrotum ( Fig. 16.8 ). Uterus and fallopian tubes are usually well formed, but occasionally may be hypoplastic. In mixed GD, the development of wolffian and müllerian structures and the virilization of external genitalia correlate with the degree of development of the ipsilateral testis resulting in asymmetric virilization of the external or internal genitalia and unilateral cryptorchidism. Pelvic ultrasound can often detect the presence or absence of male or female internal genitalia and, in the case of mixed XY GD, asymmetry in development of the müllerian and wolffian structures. There are rare reported cases of patients presenting with premature adrenarche in an otherwise unambiguous female, due to a testosterone-producing gonadal tumor.
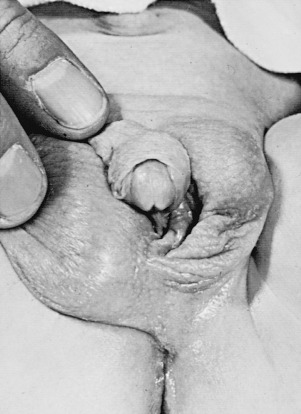
Isolated 46,XY pure GD is considered in an adolescent with primary amenorrhea and sexual immaturity with a full female external phenotype, while partial or mixed GD should be considered more likely in the differential diagnosis of a 46,XY patient with ambiguous genitalia. The biochemical changes in complete, partial, and mixed GD are outlined in Table 16.4 .
The major criteria for the diagnosis of pure, partial, or mixed GD are the appearance and histology of both gonads. Therefore, the ultimate diagnosis of any of the forms of GD, particularly the distinction between mixed and partial GD, requires biopsy of both gonads. Because the risk of gonadoblastoma in these patients is elevated, precise diagnosis of the type of GD is usually determined after prophylactic or therapeutic gonadectomy. In pure 46,XY GD both gonads are streak gonads. Partial GD is defined by bilateral dysgenetic gonads, whereas mixed GD typically has one streak gonad. Karyotype of peripheral leukocytes shows 46,XY in pure and partial GD, and mosaic 45,X/46,XY is frequent in mixed GD. However, if there is a streak gonad on one side and a normal testis contralaterally, but the peripheral karyotype is 46,XY, cryptic mosaicism can often be revealed within the gonad.
Once a presumptive diagnosis is made in 46,XY patients, FISH for SRY or for Yp can be performed. Only a minority of cases of XY GD can be explained by complete or partial SRY deletion or mutation, or SF1 mutation. The majority of patients are 46,XY and positive for SRY and SF1 . Sequencing of the SRY or SF1 open reading frames for mutations is only positive in up to 30% of 46,XY pure GD. Certain isolated and syndromic forms of XY GD can be diagnosed molecularly with cytogenetics, chromosomal microarrays, or sequencing of known genes (see Table 16.4 ). For example, 46,XY DSD in conjunction with congenital heart disease may increase the suspicion for a GATA4 mutation. However, next-generation sequencing is now available in the clinical realm to replace this sequential guesswork and should be prioritized as a diagnostic tool to identify etiology.
In addition, whole-genome chromosomal microarray studies have identified many rare copy number variants associated with patients with 46,XY GD and 46,XX testicular and ovotesticular DSD, including deletions and duplications in and around SOX3 , GATA4 , WWOX , and DMRT1 . Deletions or duplications in the upstream promoter region of SOX9 are associated with isolated and familial cases of 46,XY GD and 46,XX testicular DSD.
If a mutation or translocation involving SRY is found, the father should be tested for a possible familial mutation because SRY mutations can result in a full spectrum of phenotypes, from 46,XY fertile males to ambiguous individuals to females with partial and pure GD. Due to potential autosomal modifiers, it is very difficult to predict the risk of recurrence of a GD phenotype in XY offspring.
Management of XY Gonadal Dysgenesis
The major concern in the treatment of patients with XY GD is the risk of gonadoblastoma, a mixed germ-cell, sex-cord tumor. The risk of gonadoblastoma formation in XY GD patients increases with age and has been estimated to be as high as 30% by 30 years of age. Due to the high risk of gonadoblastoma formation, patients typically undergo prophylactic or therapeutic gonadectomy in the first decade of life. Patients are followed with regular ultrasounds every 6 months starting at age 2 until gonadectomy can be performed. With the advent of laparoscopic techniques, the invasiveness of these surgeries is decreasing and the results are improving.
In patients with XY pure GD, genitals and gender identity are unambiguously female. There are no reported cases of gender dysphoria. In patients with partial or mixed GD, the degree of genital masculinization can be used for initial gender assignment. However, issues of gender identity do arise in partial and mixed GD and are outlined further in the management section.
To complete secondary sex development, sex steroid replacement should be initiated at puberty. Sex steroid replacement is essential not only for development of secondary sex characteristics, but also growth spurt and normal accrual of bone mineral density. Height, weight, and bone density should be monitored regularly. Furthermore, a dual-energy x-ray absorptiometry (DEXA) scan for bone density should be performed prior to induction of puberty with exogenous hormone replacement, and yearly thereafter for the first 2 years. As long as the first three DEXA scans are reported as normal, they can be done every 2 to 3 years.
Because a normal uterus is often present, hysterectomies are not common to preserve childbearing potential with in vitro fertilization methods. Despite this, most patients do not carry successful pregnancies for unknown reasons. Mixed XY GD individuals with more masculinization of the external genitalia are often raised as males and consequently require lifetime testosterone therapy, through intramuscular injections or transdermal patches or gel.
Differential Diagnosis: Testicular Regression Syndrome
Testicular regression syndrome (TRS), also known as vanishing testes , results from an insult after initial testes determination, most likely vascular thrombosis or testicular torsion. Internal and external genitalia are variably developed, which presumably depends on when the loss of fetal testes occurred. Rarely, patients can present in adulthood as females with primary amenorrhea. Typically TRS is characterized by primitive epididymis and spermatic cord in the presence of a fibrous nodule with hemosiderin deposition rather than the streak or dysgenetic gonads seen in 46,XY GD. The presence of spermatic cord structures, the absence of müllerian derivatives, and typical male external phenotype suggest viable testes existed early in development, and imply a late fetal or early neonatal regression. Diagnostic criteria are outlined in Table 16.4 .
The incidence of TRS has been estimated at 5% of males presenting with cryptorchidism and as high as 12% of cryptorchid patients older than 1 year. Correct diagnosis of TRS versus 46,XY GD is essential because of significant malignant potential of abdominal or dysgenetic testes. Mutational analysis of SF1 as a cause of TRS did not find any association between mutations in SF1 and TRS and the etiology of TRS remains unknown. Although the condition is thought to be rather frequent, optimal management remains unclear. No reports of testicular germ cell tumors in TRS have been published, suggesting that excision of the remnant gonad may be unnecessary.
Disorders of Sex Differentiation: 46,XY Disorders of Androgen Biosynthesis
46,XY DSD, with phenotypes ranging from typical female to undervirilized XY males, can also be a result of defects in steroidogenic enzymes ( Fig. 16.9 ) (see Chapter 4 ). The biosynthesis of testosterone is essential for development of secondary sex characteristics in XY individuals, including differentiation of wolffian structures, inguinoscrotal testes descent, and masculinization of external genitalia after conversion to DHT. Additionally, mutations in Sertoli cell products, such as AMH or its receptor, can result in incomplete regression of female internal genitalia. Mutations in the synthesis and action pathways of testosterone, or upstream regulators of testosterone production, can all cause incomplete virilization or müllerian regression in the XY male. However, in these individuals, the testes are of normal size.
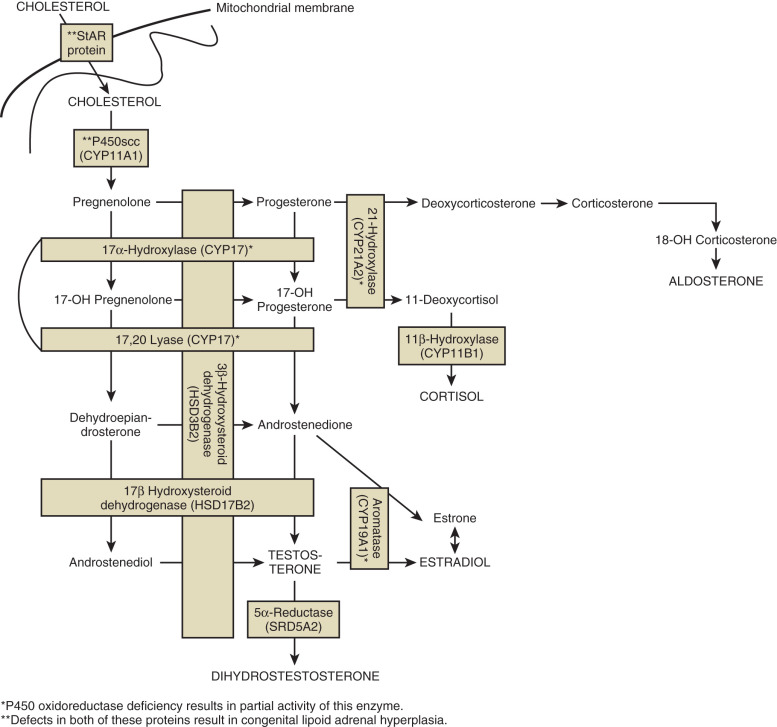
Congenital Lipoid Adrenal Hyperplasia
Mutations in genes encoding proteins involved in the initial enzymatic regions of steroidogenesis, such as steroidogenic acute regulatory protein (StAR) and cytochrome P450 side chain cleavage (P450scc, also known as CYP11A1) , result in global silencing of adrenal and gonadal steroidogenesis. The accumulation of cholesterol in the adrenals and gonads ultimately results in primary adrenal and gonadal failure. StAR protein transports cholesterol across the inner and outer mitochondrial membranes of steroidogenic cells. P450scc catalyzes the initial reaction in all steroidogenic tissues, the conversion of cholesterol to pregnenolone (see Fig. 16.9 ). On adrenal histology, patients with mutations in StAR protein or P450scc have lipid vacuoles. However, patients with STAR mutations have adrenal hyperplasia, whereas patients with P450SCC mutations typically do not exhibit adrenal enlargement.
Presentation and Diagnosis of Congenital Lipoid Adrenal Hyperplasia
Congenital lipoid adrenal hyperplasia (lipoid CAH) usually presents with a salt-wasting adrenal crisis at birth, which is usually fatal if not immediately diagnosed and treated; however, there are reports of delayed presentation. All XY patients present with a female phenotype or, in cases of partial defect, some degree of genital ambiguity. XX patients only exhibit the features of adrenal insufficiency at birth.
46,XY patients have testes, no müllerian structures due to the presence of AMH, partial or absent wolffian derivatives due to a lack of testosterone biosynthesis, and a blind vaginal pouch. The spectrum of external genital phenotype ranges from female to ambiguous external genitalia. Testes can be in the abdomen, inguinal canal, or labia. Additionally, intrauterine glucocorticoid deficiency results in elevated adrenocorticotropic hormone (ACTH) levels, which manifests clinically as generalized hyperpigmentation at birth.
Partial deficiency of StAR protein may, in 46,XX patients, result in spontaneous puberty, menarche, and anovulatory menses because their ovaries are able to produce estrogen through StAR-independent pathways. However, at puberty, XX females develop multiple cysts in their ovaries, possibly from anovulation. Because the ovaries do not produce steroids until puberty, they are spared from the cholesterol-induced damage, which occurs in the adrenals from birth.
Definitive diagnosis of congenital lipoid adrenal hyperplasia is done by sequencing of StAR or CYP11A1 . Characteristic biochemical abnormalities are outlined in Table 16.4 .
Management of Congenital Lipoid Adrenal Hyperplasia
Patients should be given physiologic replacement doses of glucocorticoid and mineralocorticoids. At the onset of puberty, patients should be given sex hormones in concordance with phenotypic sex.
3β-Hydroxysteroid Dehydrogenase Deficiency
This rare variant of CAH can have a wide spectrum of phenotypes, from classical presentation (salt-wasting, with a range of female external genitalia to ambiguous genitalia in genetic XY individuals) to nonclassical (no salt-wasting and later presentation). Rare defects in adrenal and gonadal 3β-hydroxysteroid dehydrogenase (HSD3B2) affect the synthesis of three major adrenal steroid hormones: cortisol, aldosterone, and testosterone synthesis. The resulting phenotype is adrenal insufficiency in XX and XY patients but genital ambiguity only in XY patients.
17α-Hydroxylase/17,20-Lyase Deficiency
Biosynthesis of adrenal and sex hormones requires cytochrome P450 17α-hydroxylase/17,20-lyase (P450C17) encoded by the CYP17A1 gene. Here, a single protein, P450C17, catalyzes two steps in the steroidogenic pathway, both of which are essential for sex steroid and glucocorticoid synthesis. Mutations in this gene ultimately result in decreased cortisol synthesis with shunting of steroidogenesis toward a mineralocorticoid precursor that has mineralocorticoid activity (see Fig. 16.9 ). Isolated cases of 17,20-lyase deficiency are caused by mutations in Cytochrome b5 (CYB5A), a gene that promotes the specific allosteric interaction between CYP17A1 and POR.
Presentation and Diagnosis of 17 α -Hydroxylase/17,20-Lyase Deficiency
Most patients present at puberty with primary amenorrhea, low-renin hypertension, and hypokalemia (due to elevated mineralocorticoids). This autosomal recessive disorder results in ambiguous genitalia only in 46,XY patients. 46,XY-affected individuals have phenotypes ranging from female external genitalia with no wolffian duct development to ambiguous genitalia with partial development of the male ductal system, depending on the severity of the enzymatic defect. All patients show müllerian structure regression, due to the presence of functional Sertoli cells. In contrast to individuals with 3βHSD deficiency or lipoid CAH, patients do not develop adrenal crisis due to the presence of glucocorticoid precursors that maintain some activity. Serum diagnostic criteria are outlined in Table 16.4 . Purely endocrine diagnosis can be ambiguous and definitive diagnosis is by sequencing of the CYP17A1 gene. Rare CYP17A1 mutations that cause isolated 17,20-lyase deficiency only affect sex steroid biosynthesis and result in variably feminized 46,XY individuals with normal levels of cortisol.
Management of 17 α -Hydroxylase/17,20-Lyase Deficiency
Patients should be treated with physiologic levels of glucocorticoids and appropriate sex steroid replacements to develop secondary sex characteristics.
P450 Oxidoreductase Deficiency
Cytochrome POR deficiency is a recently characterized cause of CAH that affects the enzymatic activity of all microsomal P450 enzymes, including the steroidogenic enzymes P450c17, P450c21, and P450aro. As POR is a flavoprotein required for the action of all microsomal p450 enzymes (see Fig. 16.9 ), POR deficiency causes partial enzymatic activity loss in multiple enzymes. Therefore, POR deficiency presents with a wide range of phenotypes and is particularly difficult to diagnose using serum hormone levels and the definitive diagnosis is made by gene sequencing. Partial enzymatic deficiencies in 21α-hydroxylase account for the CAH phenotype and virilization in XX individuals, while partial 17α-hydroxylase deficiency accounts for undervirilization of XY individuals. Aromatase deficiency secondary to POR deficiency is the likely cause of virilization of mothers carrying fetuses with POR deficiency.
In addition to the genital phenotype, some POR deficiency patients present with a complex skeletal phenotype known as Antley-Bixler syndrome (ABS). These skeletal malformations include, but are not limited to, craniosynostosis, radiohumeral synostosis, midface hypoplasia, and choanal stenosis.
Presentation and Diagnosis of Oxidoreductase Deficiency
The unique pathophysiology of POR deficiency results in a wide phenotypic spectrum, in which XX and XY patients present in the prenatal period, as newborns with ambiguous genitalia, or as XX adolescents with amenorrhea and polycystic ovarian syndrome (PCOS). During prenatal screening, this diagnosis should be considered in the differential of a pregnant woman with low estriol levels but with a normal ultrasound exam and virilization. Endocrine changes are shown in Table 16.4 . The majority of patients have compound heterozygous mutations in POR . The most common mutation is Ala287Pro in Caucasians, and Arg457His in Japanese patients. More severe genital and skeletal malformations are associated with loss of function alleles, while missense mutations result in mild to moderate genital and skeletal malformations.
17β-Hydroxysteroid Dehydrogenase Deficiency
The17βHSD3 enzyme is a member of the 17β-HSD family, and catalyzes the conversion of Δ4A to testosterone in the testes (see Fig. 16.9 ). This is a rare cause of 46,XY DSD, with less than 30 reported cases in the literature.
Presentation and Diagnosis of 17 β HSD3 Deficiency
In XY individuals with female external genitalia, HSD17B3 deficiency may be difficult to recognize in infancy because they manifest no other significant clinical problems and have almost normal lab values. However, depending on the degree of residual enzyme activity, HSD17B3 deficiency can cause a range of undervirilization of XY individuals at birth, from an empty bifid scrotum with perineoscrotal hypospadias to clitoromegaly and posterior labioscrotal fusion. Testes can be undescended or labial. Müllerian ducts are absent but interestingly, wolffian duct derivatives are present, despite the decreased testosterone production. During puberty, 46,XY patients may experience progressive adrenarche, including virilization, clitoral-phallic enlargement, muscle and breast development, testicular descent, and plasma T levels within the normal range of pubertal males. To date, no affected 46,XX females have been reported. Biochemical changes in patients with HSD17B3 mutations are outlined in Table 16.4 .
Management of 17 β HSD3 Deficiency
XY patients can be raised as females or males. However, in one study, up to 40% of affected XY individuals who were raised as female successfully switched gender roles at puberty. General management is covered later in this chapter.
5α-Reductase Type 2 Deficiency
SRD5A2 deficiency results from a defect in the enzyme that converts testosterone to DHT. DHT is responsible for the differentiation of male external genitalia (see Fig. 16.2 ). Peripheral conversion of testosterone to DHT is an irreversible reaction catalyzed by the two isoenzymes of 5α-reductase, SRD5A1 and SRD5A2. During puberty, the SRD5A1 isozyme is active in skin fibroblasts. During fetal development, the SRD5A2 isozyme is expressed in the genital skin tissue and male accessory sex organs. Mutations in SRD5A2 result in undermasculinized external genitalia (e.g., micropenis, perineal hypospadias). Examination of internal genitalia demonstrates normally differentiated wolffian ducts, with no müllerian structures, in addition to a blind vaginal pouch. Adrenal and gonadal biosynthesis remains normal.
Presentation and Diagnosis of 5α-Reductase Type 2 Deficiency
In 46,XY infants with mutations in SRD5A2 , the degree of ambiguity ranges from isolated hypospadias to severe undermasculinization, complete with perineal hypospadias, micropenis, bifid scrotum, and hypoplastic prostate. At puberty, 46,XY patients experience a surge of testosterone, which can cause development of male secondary sex characteristics, such as male body habitus, deepening of the voice, and penile enlargement. However, these individuals have unusually fine upper lip hair. Due to the underdevelopment of the prostate and seminal vesicles, semen is highly viscous and ejaculate volume is extremely low (<1 mL), although sperm counts are normal. Patients who are unambiguously female at birth often present in puberty with complaints of amenorrhea, a deepening voice, clitoromegaly, and increased musculature. In contrast to patients with 17βHSD3 deficiency and AIS, mutations in SRD5A2 do not cause gynecomastia during puberty. 46,XX individuals with SRD5A2 deficiency have typical female sexual differentiation and fertility, but delayed puberty and sparse pubic hair.
Diagnosis of SRD5A2 deficiency during early infancy and puberty can be made based on an elevated ratio of testosterone to DHT (normal <30 : 1), either with or without human chorionic gonadotropin (hCG) stimulation. DHT is low in infants but can reach near-normal levels during adolescence without treatment, presumably by peripheral SRD5A1 activity. If the diagnosis is suspected during prepubertal childhood or during adolescence, hCG stimulation is necessary to elicit the diagnostic hormone levels because the hypothalamo-pituitary-gonadal axis is inactive. Affected XX individuals are phenotypically normal, but have the same biochemical abnormalities (outlined in Table 16.4 ) as affected XY patients.
Definitive diagnosis is through sequencing of the SRD5A2 gene. Homozygous or compound heterozygous mutations have been reported in all five exons of the gene. To date, over 50 mutations in SRD5A2 have been found, ranging from point mutations resulting in missense or nonsense mutations, deletions, and uniparental isodisomy.
Management of 5α-Reductase Type 2 Deficiency
This autosomal recessive disorder has been described in a number of consanguineous families in Turkey, New Guinea, Saudi Arabia, and the Dominican Republic. Many XY patients with this disorder are raised as female during childhood, but upon the onset of puberty, identify with the male gender. It is not clear why there is such a high prevalence of change in gender identity in this disorder, but it does not appear to be related to the degree of external genitalia masculinization at birth. Topical DHT cream application in the pubic area promotes phallic growth prior to puberty and local application results in increased facial and body hair. These patients are not spontaneously fertile despite their normal sperm counts due to their highly viscous ejaculate. However, fertility is possible with surgical correction of the male ductal system or through assisted reproductive technology.
“Backdoor” Pathway to Fetal Testicular Androgen Production
An alternative pathway toward androgen synthesis, via the conversion of 17-OH progesterone to DHT, has been described in familial and isolated human patients with 46,XY DSD. The number of allelic mutations in the two genes AKR1C2 and AKR1C4 determine the severity of the genital phenotype.
Isolated 46,XY Disorders/Differences of Sex Development With Apparently Normal Testosterone Levels
Mastermind-like domain containing 1 (MAMLD1) is expressed during fetal Leydig cell development and important in modulation of fetal testosterone synthesis. Nonsense mutations in MAMLD1 have been identified in several patients with isolated 46,XY DSD with severe hypospadias and microphallus. Mutations in MAMLD1 result in a decrease in MAMLD1’s transactivating properties and a reduced testosterone and CYP17A1 expression in vitro. Specific polymorphisms in MAMLD1 have also been associated with hypospadias.
46,XY Disorders of Androgen Action
Partial and Complete Androgen Insensitivity Syndrome
In the presence of normal gonads and steroid biosynthesis, mutations in the steroid hormone receptors can also mute the effects of circulating steroid on specific tissues. Mutations in the AR, which is located on the X chromosome, result in AIS. Both testosterone and DHT can bind to AR and transcriptionally activate downstream genes. AR plays important roles in the differentiation of male internal and external genitalia and in the maintenance of spermatogenesis. Currently, there is an estimated incidence of 1 : 20,400 live-born XY individuals, in whom more than 300 mutations have been identified.
AIS is characterized by a range of phenotypes in 46,XY individuals, from unambiguous XY female, termed complete AIS (CAIS), to phenotypic XY males. In CAIS, the major reproductive manifestation is amenorrhea, typically presenting at puberty, and the associated infertility. At the other extreme is minimal AIS (MAIS) in which phenotypically male patients often present with infertility, gynecomastia, or hypospadias. Patients who exhibit various degrees of genital ambiguity are referred to as PAIS. CAIS is generally associated with a complete absence of androgen binding and AR activation. Beyond this, there is almost no correlation between the percentage of residual AR activity and phenotype, suggesting that other genetic modifiers can modulate the phenotype. Even within families, the phenotype resulting from the same mutation can vary between PAIS and CAIS.
The majority of mutations resulting in AIS are caused by single amino acid substitutions, which account for approximately 90% of the cases. Mutations are found throughout the gene, with the majority of mutations located within the ligand-binding domain. Exon 1 rarely possesses a causative mutation. The majority of cases are inherited, but 30% of all AIS cases are de novo mutations, which is consistent with other X-linked traits.
In familial PAIS, and in both familial and sporadic CAIS, mutations in AR exonic sequences are found in 85% to 90% of cases. In contrast, detectable mutations in AR account for only 10% to 15% of sporadic cases with hormonal profile and clinical presentation suggestive of de novo PAIS, and the molecular defect in these cases is unknown. Although the genotypes causing CAIS are consistent in phenotypic presentation, in PAIS there is phenotypic variability among affected individuals carrying the same mutation, and many individuals carrying a working diagnosis of PAIS will not have AR mutations. The term PAIS should be reserved for individuals in whom causative AR mutations have been found.
Presentation and Diagnosis of Androgen Insensitivity Syndrome
The phenotype of patients with PAIS is extremely heterogeneous. Patients present in infancy or childhood with variable degrees of virilization such as micropenis, cryptorchidism, and perineoscrotal hypospadias. Infants and children may present with unilateral or bilateral inguinal hernias. Testes are present and functional, producing high levels of testosterone and AMH resulting in variable wolffian derivative development and regression of müllerian structures in most patients. In patients presenting at puberty, breast development and sparse pubic hair are suggestive of PAIS, and help differentiate it from an SRD5A2 deficiency.
CAIS usually presents at puberty with primary amenorrhea. Physical exam reveals a short, blind vagina, absent uterus, and sometimes palpable inguinal or labial testes. Testosterone-dependent wolffian derivatives and prostate are absent or vestigial. Height, bone maturation, and breast development are normal but pubic and axillary hair, an androgen-mediated feature, is absent or sparse. Patients’ identity and behavior are feminine without gender dysphoria. Less commonly, CAIS may present in infancy with phenotypic female genitalia and inguinal or labial masses representing testes.
Diagnostic criteria for serum hormone levels are outlined in Table 16.4 . In prepubertal children, basal luteinizing hormone (LH) and testosterone may be normal, but hCG stimulation elicits an exaggerated androgen response (a tripling instead of a doubling of testosterone and DHT). Exam and pelvic ultrasound reveals abdominal testes and the absence of müllerian structures.
Molecular genetic testing of the AR gene detects mutations in more than 95% of probands with CAIS. Molecular testing has been shown to be more consistent than biochemical functional assays of AR function, which has been discredited based on the high degree of variation due to biopsy site and testing laboratory. PCR-based sequencing of AR exons 2 to 10 can be routinely performed, as well as sequencing of the much longer exon 1, and some intronic and promoter regions. Deletion/duplication testing of the AR gene is also available. Prenatal testing by mutation analysis is available for families in which the AIS-causing allele has been identified in an affected family member.
Luteinizing Hormone Receptor Defects
Leydig Cell Hypoplasia
Leydig cell hypoplasia (LCH) or aplasia is a disorder caused by inactivating mutations of the joint G protein-coupled receptor for human luteinizing hormone and choriogonadotropin (LHCGR). It is characterized by impaired Leydig cell differentiation and testosterone production. In utero, placental hCG stimulates Leydig cells to produce testosterone, resulting in male internal and external genitalia. LH takes over during the third trimester of gestation and neonatal life to complete Leydig cell development and continue testosterone production.
Over 20 inactivating mutations of LHCGR have been identified, scattered throughout the gene, that cause variable degrees of loss of receptor activity. More severe mutations resulting in truncation, decreased surface expression, or decreased coupling efficiency are usually associated with unambiguous female phenotype. Partial inactivating mutations often result in an undervirilized phenotype such as micropenis or hypospadias. One specific mutation highlights the differential binding sites of hCG and LH to the same receptor. A splice variant resulting in the absence of exon 10 was described in an 18-year-old male who presented with normal male phenotype, pubertal delay, small testicles, and delayed bone age. The receptor’s response to hCG was normal, inferred from normal fetal sex development. However, LHCGR was not activated by LH, resulting in delayed puberty and bone age.
Presentation and Diagnosis of Leydig Cell Hypoplasia
Patients with severe inactivating mutations in LHCGR are often missed at birth and present in puberty with amenorrhea. Partially inactivating mutations present at birth, with undervirilization of the external genitals (e.g., micropenis, hypospadias, and cryptorchidism). In XX females, LHCGR mutations result in hypergonadotropic hypogonadism with primary amenorrhea or oligomenorrhea, cystic ovaries, and infertility. However, XX females will undergo spontaneous breast and pubic hair development during puberty.
Pelvic ultrasound demonstrates absence of uterus and fallopian tubes. To distinguish LCH from patients with XY GD, AMH is used as a marker of testicular Sertoli cell function and is normal to high in LCH patients and low to undetectable in XY GD patients. Histologic analysis of the testis shows normal Sertoli cells, but hyalinized seminiferous tubules, without mature Leydig cells or spermatogenesis. Patients with partially inactivating mutations may show some early spermatogenesis, with arrest prior to spermiation. Therefore, patients have potential for fertility using assisted reproductive technology.
Biochemical changes are outlined in Table 16.4 . Definitive diagnosis requires sequencing of the LHCGR gene for deletions, insertions, and point mutations.
Disorders of Anti-Müllerian Hormone or Anti-Müllerian Hormone Receptor
Persistent Müllerian Duct Syndrome
During the critical time in sex differentiation, between weeks 10 and 12 of gestation, testicular AMH causes the regression of the müllerian derivatives. Inactivating mutations in AMH or AMH receptor type II (AMHR2) in XY individuals can prevent this regression, resulting in both male and female internal genitalia, with typical male external genitalia.
Presentation and Diagnosis of Persistent Müllerian Duct Syndrome
The diagnosis of persistent müllerian duct syndrome (PMDS) is commonly made incidentally during abdominal imaging studies or surgical exploration of the abdomen. XY patients with PMDS are phenotypically male. Aside from having a uterus and fallopian tubes in addition to male internal and external genitalia, they often have no clinical abnormalities. Therefore the true prevalence of PMDS is difficult to ascertain. The vast majority of cases of PMDS have mutations in the AMH or AMHR2 in approximately equal proportions. Inheritance is autosomal recessive for both loci.
Boys can also present with cryptorchidism (20%) or with an inguinal hernia containing müllerian structures, but normal virilization. The increased likelihood of abdominal testes in PMDS causes an increased incidence of gonadoblastoma.
Related posts:
 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Hormone-Responsive Cancers
Hormone-Responsive Cancers
 Medical Approaches to Ovarian Stimulation for Infertility
Medical Approaches to Ovarian Stimulation for Infertility
 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


