THE CONNECTIVE TISSUE AND THE EXTRACELLULAR MATRIX
The connective tissue is the most abundant tissue throughout the body and serves both as a structural support and as a metabolic reservoir for tissue interactions. During recent years, the historical view on connective tissue as solely a supportive structure has been abandoned. It has become evident that the connective tissue actively participates in several important physiologic processes, including cell expression patterns and development, proliferation, differentiation, cell migration, and apoptosis, mainly acting through various cell signaling pathways. Three main connective tissues can be recognized: the loose or soft connective tissues (that embed organs and organ parts), the hard connective tissues (cartilage and bone), and blood. They consist of an extracellular matrix (ECM) and metabolically active cells that synthesize and maintain the ECM. Embryologically, connective tissue develops mainly from the mesoderm, the middle layer of the three-layered embryo. Mesodermal cells develop a loosely organized embryonic connective tissue, called mesenchyme, and differentiate further in specific cell types, including fibroblasts/fibrocytes, chondroblasts/chondrocytes, and osteoblasts/osteocytes. These cells synthesize and maintain the ECM in, respectively, the loose connective tissues, cartilage, and bone, according to the tissue’s specific needs. The ECM consists of four main components: elastic fibers and microfibrils, collagen fibers, proteoglycans, and glycoproteins.
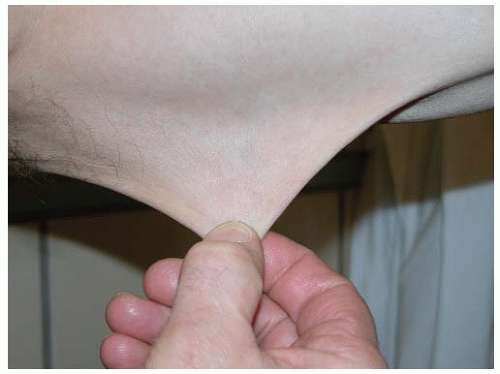
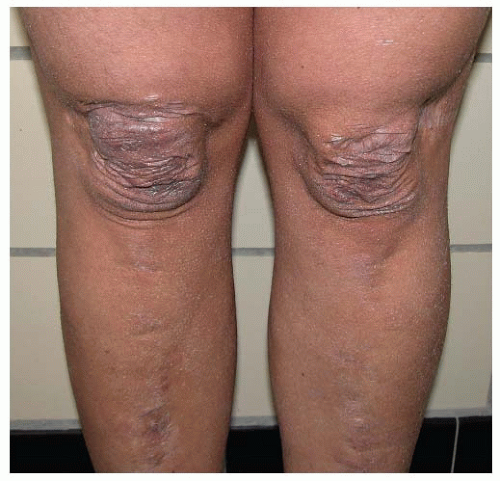
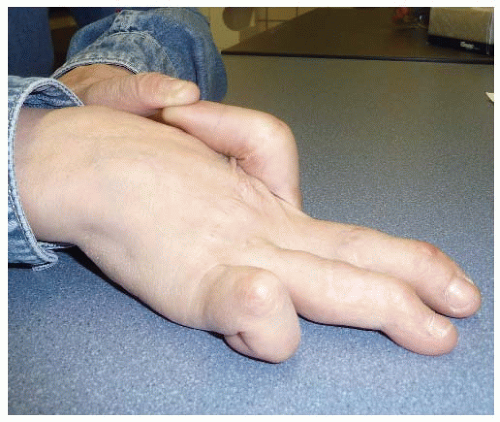
Elastic fibers provide elasticity and resilience to all deformable tissues. They are abundantly present in the aorta, arterial vessels, lung, and, to a lesser degree, skin. They are composed of two ultrastructurally distinguishable components: an amorphous component, “elastin”, which contributes to the elasticity of the fiber, and a surrounding sheet of microfibrils. The microfibrillar component of the elastic fibers consists of fibrillin-rich microfibrils. Fibrillins are highly homologous glycoproteins, of which five isoforms have been described with the same overall arrangement of repetitive domains. Fibrillin-rich microfibrils interact with many other nonfibrillar components, which can be integral parts of the microfibrils or may associate with them. These microfibrils have a structural role and are functionally important in different processes such as microfibril assembly, elastogenesis, interactions with other ECM proteins, anchoring microfibrils to basement membranes and cell surfaces, and growth factor sequestration. For many of these molecules, however, the exact spatiotemporal relation and function still need to be addressed. Examples of nonfibrillar components include microfibril-associated glycoproteins, fibulins (a seven-member family of extracellular glycoproteins), and the latent transforming growth factor-beta-binding proteins (LTBPs), which are important players in the bioavailability of transforming growth factor-beta (TGF-β) in the ECM.
Collagens are the most abundant proteins in the body. They are trimeric proteins that are characterized by the presence of triple-helical domains. To date, 43 collagen genes have been described, the products of which combine to form at least 28 different collagen molecules. Most collagens form supramolecular assemblies, such as fibrils and networks, and the collagen superfamily has been divided in several subfamilies on the basis of their genomic structures, their assembly, and other structural features. The fibril-forming or fibrillar collagens, which include the collagen types I, II, III, V and XI, represent the most widespread and abundant class of collagens, and defects in these proteins are found in different heritable disorders of connective tissue (HCTD), such as osteogenesis imperfecta (OI), (type I collagen), Stickler syndrome (type II and XI collagen), and the Ehlers-Danlos syndromes (EDS) (type I, III, and V collagen). Fibrillar collagens are observed in tissues as long, highly ordered fibrils with a characteristic banding pattern. Type I collagen, a heterotrimer of two
α1-chains and one
α2-chain, is the major collagen type in the body and has a widespread tissue distribution. Type II and type XI collagens are predominantly found in cartilage. Type III collagen, a homotrimer consisting of three identical
α1-chains, is an essential component of stretchable tissues such as the blood vessel walls, the gastrointestinal tract, the uterus, and the skin. Type V collagen is a minor fibrillar collagen that coassembles with type I collagen, and acts as a regulator of collagen fibril diameter through the retention of a noncollagenous amino-terminal domain of the pro
α1(V) collagen chain.
1 Each fibrillar collagen has a central uninterrupted triple-helical domain with short nonhelical domains at the carboxy- and amino-terminal ends. The presence of glycine, the smallest amino acid, in every third position of each chain is a prerequisite for the formation of a stable collagen helix. The biosynthesis of fibrillar collagens in the fibroblast is a complex process and starts with the synthesis of soluble precursor molecules, procollagens. These contain globular amino- and carboxy-terminal propeptide extensions, called the
N– and the
C-propeptide. Intracellular association of three pro
α chains occurs through interaction and disulphide bonding at the
C-propeptide. In this way, correct alignment of the growing polypeptide chain is obtained as required for formation and propagation of the triple helix from the
C– to the
N-terminal end of the molecule. During helix propagation, the pro
α chains undergo extensive enzymatic modifications (e.g., hydroxylation of prolyl and lysyl residues), which cease when the helix is formed. Mature triple-helical procollagen molecules are secreted into the extracellular
environment where they are converted to collagen by enzymatic removal of the
N– and the
C-propeptides. Individual collagen molecules spontaneously assemble in a nonenzymatic process to form fibrils and fibers, which are stabilized by covalent cross-linking.
Proteoglycans consist of a core protein to which short oligosaccharides or longer chains of glycosaminoglycans (GAGs) are covalently attached. There is a large variety of proteoglycans based on the types and lengths of GAGs as well as the sequence and length of the protein core. Chondroitin sulfate, keratan sulfate, heparan sulfate, and dermatan sulfate are four different forms of GAGs that consist of repeating disaccharide units. The sulfates that are present on the GAGs create a highly negatively charged environment that is hydrophilic. In addition to water, proteoglycans also bind cationic proteins, such as growth factors, resulting in a mechanism by which tissues can store growth factors in the ECM.
A large group of noncollagenous matrix glycoproteins make up a substantial part of the ECM. Important members of this group include thrombospondins (TSP), fibronectin, tenascins, laminin, and osteopontin. As “ground substance” of the ECM, they display a variety of functions in tissue morphogenesis and remodeling, including cell adhesion, cell cycle regulation, cell-matrix interactions, and interactions with growth factors and various other matrix molecules.
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



