The last two decades have seen great progress in our knowledge of the genetic and biochemical control of mineral metabolism. Moreover, our understanding of vitamin D action continues to extend well beyond the conventional roles in bone and mineral metabolism and now includes functions as a transcriptional regulator of immune function.
Clinical application of these discoveries has led to significant and practical improvements in diagnosis and treatment of disordered bone mineral metabolism as well as analysis of bone mass in children and adolescents.
Maintenance of calcium homeostasis is a complex dynamic process involving intestinal calcium absorption and excretion, renal filtration and reabsorption, and skeletal storage and mobilization (Figure 7-1).
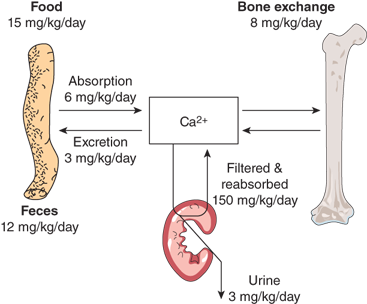
Only 1% of total body calcium is within extracellular fluids and soft tissues. Approximately 50% of total serum calcium is in the ionized form (Ca2+) at normal serum protein concentrations and represents the biologically active component of the total serum calcium concentration. Another 8% to 10% is complexed to organic and inorganic acids (eg, citrate, sulfate, and phosphate); together, the ionized and complexed calcium fractions represent the diffusible portion of circulating calcium. Approximately 40% of serum calcium is protein bound, primarily to albumin (80%) but also to globulins (20%), and is biologically inactive. Protein-bound calcium provides a readily available reserve of calcium should the acute need for increased ionized calcium arise.
Sudden changes in the distribution of calcium between ionized and bound forms may cause symptoms of hypocalcemia. Increases in extracellular fluid concentration of anions, such as phosphate, citrate, bicarbonate, or edetic acid, will decrease Ca2+ through chelation. Alkalosis increases the affinity of albumin for calcium and thereby decreases the concentration of Ca2+. By contrast, acidosis decreases the binding of calcium to albumin and increases Ca2+ concentrations.
Hence, physiologically relevant information is best obtained by determination of the Ca2+ concentration, particularly when evaluating patients who have abnormal circulating proteins or acid-base and electrolyte disorders (eg, total serum calcium will be low in a child with decreased serum albumin while the Ca2+ concentration will typically be normal). One widely used algorithm estimates that total serum calcium declines by approximately 0.8 mg/dL for each 1 g/dL decrease in albumin concentration.
The cytoplasmic concentration of Ca2+ affects many intracellular enzymes including adenylate cyclase, guanylate cyclase, troponin C, calmodulin, and protein kinase C. The normal intracellular calcium concentration can fluctuate from roughly 100 nM to greater than 1 mM, due to release from cellular stores or influx from extracellular fluid, and is generally less than the concentration in plasma (1 mM). Therefore, only minute amounts of calcium, either released from endoplasmic reticulum or from stimulated influx through calcium channels, are needed to raise the concentration by the factor of 10 that is ordinarily necessary to activate an enzyme. The activity of cell membrane calcium channels and endoplasmic reticulum ion pores is regulated by membrane potential as well as by interactions with membrane receptors and phospholipids.
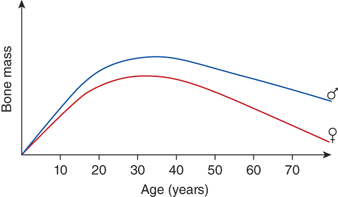
Dietary calcium requirements vary for different age groups. The primary demand for dietary calcium is to enhance bone mineral deposition. Skeletal calcium accumulates at a rate of 80 to 150 mg/day between birth and 10 years of age; this rate increases to a maximum of 200 mg/day in girls and 270 mg/day in boys during the adolescent growth spurt and gradually falls to zero with cessation of growth. Approximately 90% of the total bone mass is formed by the age of 20. The remaining 10%, which determines the maximum attainable bone mass, is accumulated during the second and third decade. Subsequently, bone mass decreases continually (Figure 7-2).
Both dietary constituents and genetic variation influence net calcium absorption. Accordingly, interactions between these factors complicate determination of a universal calcium “requirement” for all children. In response, dietary guidelines have been developed that will ensure adequate calcium intakes for most children.
Calcium absorption from the intestine is a regulated process, and is influenced by calcium intake and requirements. The efficiency of calcium absorption is much greater from the duodenum/jejunum than from the ileum. Intestinal calcium absorption occurs by two processes, a saturable, vitamin D-dependent active transport in the upper- and midsection of the small intestine and a nonsaturable, vitamin D-independent diffusional transfer along the entire small intestine. Calcium absorption is impaired by vitamin D deficiency as well as by intestinal disorders, particularly in which there is fat malabsorption with precipitation of calcium in the intestinal lumen as insoluble calcium salts of fatty acids, so-called calcium soaps. Excessive intake of fats, phosphates, phytates, and oxalates also decreases intestinal calcium absorption by formation of insoluble salts.
Renal tubular calcium reabsorption is an important determinant of extracellular fluid (ECF) calcium concentration. Normally, about 99% of filtered calcium is reabsorbed by the tubules, but the fractional excretion of calcium can be modified by active transport mechanisms that are controlled by local and systemic factors (eg, extracellular calcium, parathyroid hormone [PTH], and calcitriol). PTH and calcitriol appear to increase the activity and/or expression of transport proteins (eg, TRPV5) that increase transcellular calcium transport.1
In addition, extracellular calcium regulates its own reabsorption by the mammalian tubule. In the thick ascending limb of the distal nephron, the calcium-sensing receptor (CaSR) is involved in regulation of paracellular reabsorption of calcium and magnesium. Indeed, a large body of evidence supports the view that calcium exerts this action in almost an “autoregulatory” mechanism via activation of the CaSR. First, increasing ECF calcium concentration elicits a marked increase in urinary calcium (and magnesium) excretion—a process that occurs independently of any change in calcium-regulating hormones. Second, the inhibitory effect of ECF calcium on its own reabsorption is shared by other CaSR agonists, for example, magnesium (Figure 7-3).
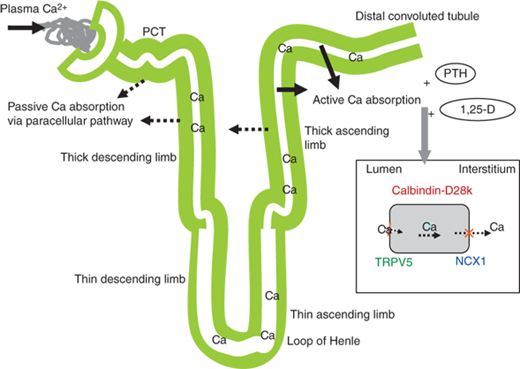
Figure 7-3
Regulation of renal calcium. Passive calcium absorption occurs in the proximal convoluted tubules (PCT) and thick ascending limb. Active calcium absorption occurs primarily in the distal convoluted tubule and collecting duct. Active absorption occurs through the permeable transient receptor potential V5 channel (TRPV5), located on the luminal membrane of the renal tubular cell. Intracellular transport occurs via calbindin-D28k and calbindin D9k. Intracellular calcium exchange with sodium to the interstitium occurs through the plasma membrane Ca2+ ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX1), located on the basolateral membrane. Active absorption is stimulated by PTH and 1,25(OH)2D (1,25-D) via increased expression of these calcium transport proteins.
Both calcium and magnesium are passively reabsorbed in the cortical portion of the thick ascending limb (cTAL) along an electrical gradient through the paracellular pathway. The electrical gradient is related to transcellular NaCl reabsorption. Chloride exits the cell across the basolateral membrane, which depolarizes the basolateralmembrane, and the overall consequence is a lumen-positive transepithelial voltage that drives calcium, magnesium, and also sodium through the paracellular pathway. The pathway permeability for calcium and magnesium requires the presence of specific proteins, claudin-16 and claudin-19,2 which are coexpressed with occludin and junctional adhesion molecule in the tight junctions of TAL. Inactivating mutations of the claudin-16 and claudin-19 genes cause a specific decrease in cTAL calcium and magnesium reabsorption and renal loss of both cations without renal sodium loss. These electrolyte losses are the hallmark of an inherited disease referred to as hypercalciuric hypomagnesaemia with nephrocalcinosis.2
The loop diuretics that specifically inhibit urinary sodium resorption (eg, furosemide) also inhibit calcium reabsorption resulting in calciuria. These diuretics can be useful adjuncts to the aggressive administration of intravenous saline in the emergency treatment of hypercalcemia. Systemic acidosis and high protein intake increase urinary calcium excretion; these effects may be exaggerated in renal stone formers.
At all ages approximately 99% of total body calcium is in the skeleton, where in combination with 89% of the total body phosphorus, it constitutes the major inorganic matrix of bone. Skeletal regulation of calcium will be described separately later in the text.
Magnesium and calcium homeostasis are interrelated, and any disturbance in homeostasis of one of these ions is commonly associated with disturbance in the other. Total body magnesium amounts to about 260 mg/kg in the infant and increases to about 330 mg/kg in adults. Sixty percent of the body’s magnesium is in bone, and most of the remaining 40% is intracellular. Only 20% to 30% of the intracellular magnesium is exchangeable, the rest being bound to proteins. The plasma concentration of magnesium is similar in infants, children, and adults, and ranges from 1.6 to 2.4 mg/dL (0.70-1.00 mmol/L), and seems to be maintained largely independently of calciotropic hormone influence. Of the total serum magnesium, approximately 30% is protein bound, chiefly to albumin, 55% is ionized, and 15% is complexed. The ionized and complexed fractions of magnesium constitute the ultrafiltrable magnesium. Similar to calcium, the concentration of ionized magnesium is influenced by serum pH, with a rise in pH leading to an increase in the proportion of protein-bound magnesium.
In children, the daily dietary requirement for magnesium ranges from 120 to 300 mg/day depending on age. Magnesium is principally absorbed in the small intestine by passive and active mechanisms that have yet to be fully delineated, but the role of melastatin-related subfamily of transient receptor potential 6 (TRPM6) has been highlighted by the identification of loss-of-function mutations in TRPM6 in patients with hypomagnesemia and secondary hypocalcemia due to magnesium malabsorption.3 About 50% to 75% of ingested magnesium is absorbed. In contrast to calcium balance, magnesium homeostasis is primarily maintained by renal mechanisms. Normally, less than 5% of the filtered magnesium is excreted. The major site of reabsorption is the TAL, which relies on the Na+–K+–2Cl− cotransporter, the renal outer medulla potassium channel, CaSR, and Ca+/Mg2+-sensing receptor. Both magnesium and calcium have the capacity to diminish the resorption of the other.
Phosphorus is a vital component of bone mineral, membrane phospholipids, nucleotides that provide energy and serve as components of DNA and RNA, and phosphorylated intermediates in cellular signaling. Plasma phosphorus exists in an inorganic form and an organic form. Approximately 20% of the plasma inorganic phosphorus is protein bound, and the remainder circulates as free phosphate ions HPO42− or H2PO4−. Phosphorus in the form of phosphate ions is filtered at the glomerulus. The concentration of plasma and urinary phosphate is typically expressed in terms of elemental phosphorus. Because organic phosphorus is in the form of phosphate, the latter term is used in this chapter.
Eighty to ninety percent of body phosphate is present in bone mineral as a major component of hydroxyapatite. The remainder is in soft tissue, blood cells, and in extracellular fluid. In cells, phosphate is bound to sugars, lipids, proteins, nucleic acids, and various nucleotides. In plasma, two-thirds of total phosphate is present as phospholipids. These compounds are insoluble in acid and are not measured in routine plasma phosphate determinations.
Physiological concentrations of phosphate in plasma vary by age (and growth) to a greater degree than those for calcium. The average plasma concentration in premature infants is 7.9 mg/dL (2.6 mmol/L), in full-term infants 6.1 mg/dL (2.0 mmol/L), in children and adolescents 4.6 mg/dL (1.5 mmol/L), and in adults 3.5 mg/dL (1.15 mmol/L). Serum phosphate concentrations exhibit a diurnal rhythm, with levels lowest in the morning and increasing during the day. Thus, it is best to monitor plasma phosphate concentrations in patients taking oral phosphate supplements in the fasting state and at a consistent time of day.
The principal sources of dietary phosphate are dairy products and meat. Recommended daily intake is 880 mg/day for children 1 to 10 years of age and 1200 mg/day for older children.
The principal organ regulating phosphate homeostasis is the kidney (Figure 7-4). Serum inorganic phosphorus (Pi) is filtered by the glomerulus, and 80% of the filtered load is reabsorbed predominantly along the proximal nephron. Regulation of proximal renal tubular reabsorption of phosphate is achieved through changes in the activity, number, and intracellular location of type 2 sodium-phosphate cotransporters (Npt2a and Npt2c). Phosphate reabsorption is a saturable process and is dependent on both glomerular filtration rate (GFR) and plasma phosphate concentration. The tubular maximum rate of phosphate reabsorption (TmP) is not an absolute constant, as it can be set at different levels according to physiological or pathological conditions. The TmP in relation to GFR (TmP/GFR) is the most reliable quantitative estimate of the overall tubular phosphate transport capacity. The TmP/GFR is 4.0 to 5.9 mg/dL in normal children between 6 and 14 years of age and declines to adult values of 2.5 to 4.2 mg/dL by the age of 20 years. PTH inhibits phosphate reabsorption by lowering the TmP/GFR via cAMP-dependent inhibition of Npt2a and Npt2c expression, resulting in phosphaturia. PTH is an important regulator of phosphate reabsorption, but its principal function is to maintain calcium homeostasis. By contrast, both acute and chronic phosphate deprivation initiates an adaptive increase in brush border membrane sodium- phosphate transport through recruitment of Npt2 proteins to the apical membrane surface.
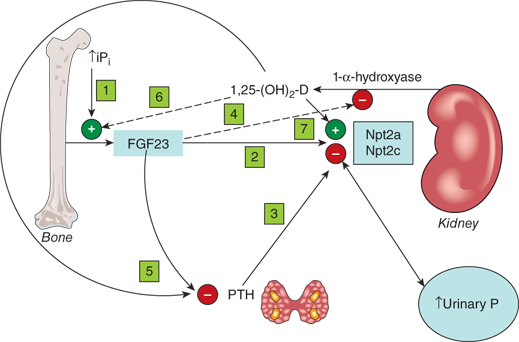
Figure 7-4
Renal regulation of phosphate. (1) Increased plasma inorganic phosphate (Pi) increases FGF23 secretion. (2) FGF23 decreases expression of renal tubule sodium-phosphate cotransporters Npt2a and Npt2c, leading to decreased Pi reabsorption. (3) PTH also decreases expression of renal Npt2a and Npt2c. (4) FGF23 decreases expression of renal 25-hydroxycalciferol 1-α-hydroxylase thereby indirectly decreasing intestinal Pi absorption. The net effect is lowering of plasma Pi. (5) FGF23, like 1,25(OH)2D, inhibits PTH secretion. (6) In contrast, 1,25(OH)2D increases FGF23 secretion, presumably offsetting 1,25(OH)2D-mediated increases in intestinal Pi absorption, and (7) likely increases Npt2a and Npt2c expression.
The primary phosphate-regulating hormones are termed “phosphatonins,” and fibroblast growth factor 23 (FGF23) is now recognized as the principal regulator of phosphate homeostasis. FGF23 reduces apical localization and expression of the renal tubular sodium- phosphate cotransporters Npt2a and Npt2c, thereby reducing phosphate reabsorption in the proximal renal tubule. In addition, FGF23 affects vitamin D metabolism by inhibiting expression and activity of the renal 1-α-hydroxylase (CYP27B1) while inducing activity of the renal 24-hydroxylase (CYP24).
Many other hormones and cytokines influence phosphate reabsorption in the proximal renal tubule. GH, IGF-I, insulin, epidermal growth factor, thyroid hormone, and dietary phosphate depletion stimulate renal phosphate reabsorption. In contrast, renal phosphate reabsorption is inhibited by parathyroid hormone-related protein (PTHrP), calcitonin, atrial natriuretic factor, TGF-α and TGF-β, and glucocorticoids.
Up to two-thirds of the dietary phosphate is absorbed from the intestine, primarily in the jejunum. The percentage of inorganic phosphate absorbed increases as inorganic phosphate intake decreases, but the absolute absorption of inorganic phosphate increases proportionally with increasing intake. Active absorption of phosphate is mediated by the type 2b sodium-phosphate cotransport (Npt2b), which is located in the brush border of enterocytes.
Dietary phosphate restriction and hypophosphatemia stimulate calcitriol synthesis via enhanced activity of the 25(OH)D-1β-hydroxylase (CYP27B1) in the kidney. This effect is mediated through inhibition of FGF23 secretion by hypophosphatemia. In turn, calcitriol activates intestinal calcium and phosphorus absorption and enhances osteoclast-dependent mobilization of calcium and phosphorus from bone. The consequent increase in serum calcium concentration, and calcitriol, together reduce PTH secretion, thereby facilitating renal excretion of excess calcium and increased reabsorption of phosphate. Ultimately, the serum calcium level is not altered and the serum phosphorus level is normalized (see Figure 7-4).
Bone mass is determined both by modeling and remodeling. Bone modeling begins as the skeleton forms during early fetal life and continues until growth ends after puberty. During this time the skeleton also undergoes continuous alteration through the process of remodeling, which shapes the bones and prevents loss of the medullary space of the long bones. Remodeling continues throughout life, and permits the skeleton to replace old or injured bone, such that the skeleton is essentially replaced every 10 to 15 years. Pubertal gain in bone mass represents the largest percentage increase after that seen in the first year of life. Once peak bone mass is achieved during the third decade of life, a period of relatively stable bone mass ensues, where bone formation and bone resorption are essentially equivalent. Bone mass begins to fall in women after the menopause, and in both genders as a function of advancing age as the rate of bone resorption exceeds the rate of formation. Peak bone mass is greatly influenced by genetic factors, and, based on studies in mono- and dizygotic twins, genetic variation may account for up to 80% of achieved peak bone mass. Environmental factors such as nutrition, exercise, hormonal milieu, and potential disease processes also influence acquisition of peak mass, and account for approximately 20% of bone mass accrual.
Structural integrity and metabolic function of the skeleton require the coordinated interaction of osteoblasts and osteoclasts and a great many systemic and local factors (Table 7-1). Osteoblasts are bone-forming cells, and are derived from mesenchymal stem cells that can also differentiate into muscle, adipocytes, cartilage, or fibrous tissue. By contrast, osteoclasts are derived from hematopoietic precursor cells that are related to macrophages.
| Bone Formation | Bone Resorption | |
|---|---|---|
| Calcium regulating hormones | ||
| PTH | (+) | + (−) |
| Calcitriol | (+) | + (−) |
| Calcitonin | − | |
| Other systemic hormones | ||
| Glucocorticoid | − | |
| Thyroid | + | + |
| Insulin | + | |
| Growth hormone | + | |
| Estrogen | + | − |
| Androgens | + | − |
| Growth factors | ||
| IGF-1 | + | |
| Epidermal growth factor | + | + |
| FGF1 | + | |
In the process of bone formation, preosteoblasts and osteoblasts synthesize and secrete type 1 collagen and the ectoenzyme alkaline phosphatase. With further differentiation, osteoblasts secrete matrix proteins, particularly osteocalcin, also termed bone gla protein due to the presence of vitamin K–dependent γ-carboxylation of glutamic acid residues. Osteocalcin is a noncollagenous bone matrix protein that is involved in the mineralization process. Osteocalcin is also released into the circulation, where its decarboxylated form appears to function as a hormone that participates in the regulation of glucose metabolism and fat mass. Cell-based studies show that decarboxylated osteocalcin can increase insulin secretion and proliferation of pancreatic β-cells, and can also enhance adiponectin secretion from fat cells. These actions suggest that osteocalcin can regulate glucose metabolism and affect insulin sensitivity and fat mass and provide evidence of a novel metabolic interaction between the skeleton and fat tissue.4 Osteoblastic activity is associated with increased serum levels of bone alkaline phosphatase, but serum osteocalcin concentrations are a more accurate indicator of bone mineralization. For example, serum osteocalcin concentrations are not elevated in children with active rickets despite greatly increased levels of bone alkaline phosphatase. The kidneys excrete osteocalcin, and serum values are increased in renal failure. Osteocalcin and other osteoblast markers are usually used to estimate the rate of bone formation, and these levels typically follow the same pattern as markers of bone resorption (see below), because the two processes are coupled during normal bone remodeling. Concentrations of osteocalcin are greatest during infancy and at puberty, and in general levels parallel the normal pattern of growth and puberty. After adolescence, bone remodeling slows, and the concentrations of these markers decline.
Fully differentiated osteoblasts are spatially arranged on the bone surface so that the deposition of collagen and noncollagen proteins occurs in a manner that is favorable for mineralization. Mineralization is delayed for several days, during which time the collagen fibrils are cross-linked to enhance strength.
As osteoblast production of collagen and noncollagen protein is completed, some osteoblasts find themselves buried within the matrix and become osteocytes. Elongated nerve processes stretch through canaliculi within the bone to connect the osteoblasts and osteocytes. This syncytium of interconnected cells forms a “mechanostat” that functions to sense mechanical load and other forces that are applied to the skeleton. Increased mechanical load stimulates osteoblastic bone formation, whereas decreased mechanical load is associated with increased osteoclastic bone resorption.
Osteoblasts express specific receptors that recognize local and systemic factors that influence bone remodeling, and they produce many regulators of bone growth. These receptors include those for PTH, calcitriol, glucocorticoids, sex hormones, growth hormone, (GH), and thyroid hormone. Moreover, receptors for interleukins such as IL-1 and IL-6, tumor necrosis factor-alpha (TNF-α), prostaglandins, insulin-like growth factors (IGFs), transforming growth factor (TGF)-β, bone morphogenetic proteins, fibroblast growth factors, and platelet-derived growth factor are also present. Factors produced by osteoblasts that probably act locally include prostaglandins, IL-6, IGFs and their binding proteins (IGF-BPs), TGF-β, bone morphogenetic proteins, fibroblast growth factors, platelet-derived growth factor, and vascular-endothelial growth factor.
Bone is resorbed by osteoclasts, large, multinucleated cells that can dissolve mineral and degrade matrix. Osteoclasts are related to monocyte/macrophage cells and are derived from granulocyte/macrophage-forming colony units (CFU-GM). Osteoclast progenitors are present in the marrow, spleen, and in small numbers in the circulation. During development, osteoclast precursors probably migrate to the bone from extramedullary sites of hematopoiesis. The lifespan of an osteoclast may be as long as 3 to 4 weeks.
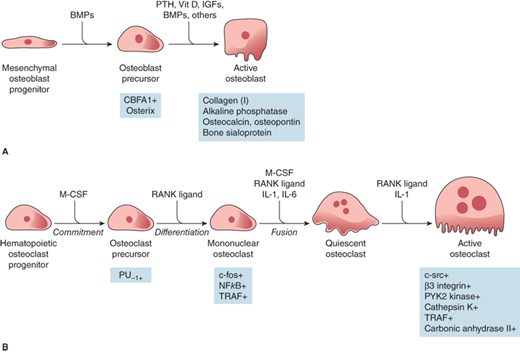
Osteoclast formation requires an interaction with cells of the osteoblastic lineage, which may depend on cell-cell contact and specific cytokines of the receptor activator of NF-kappa B (RANK) system described in the section “Resorption.” Osteoblasts may also be the source of the M-CSF that is required for osteoclastogenesis (Figure 7-5). Excessive osteoclastic resorption occurs in osteoporosis, Paget disease, hyperparathyroidism, and inflammatory bone loss. On the other hand, osteoclastic resorption is deficient in osteopetrosis.
Figure 7-5
Regulation of osteoblast (A) and osteoclast (B) development. Hormones, cytokines, and growth factors regulating cell proliferation and differentiation are shown above the arrows. Transcription factors regulating cell proliferation and differentiation are shown below the arrows. BMPs, bone morphogenic proteins; PTH, parathyroid hormone; Vit D, vitamin D; IGFs, insulin-like growth factors; CBFA1, core binding factor A1; M-CSF, macrophage colony-stimulating factor; PU-1, a monocyte- and B lymphocyte-specific ets family transcription factor; NFkB, nuclear factor kB; TRAF, tumor necrosis factor receptor-associated factors; RANK ligand, receptor activator of NFkB ligand; IL-1, interleukin 1; IL-6, interleukin 6. (Reproduced with permission from Jameson JL. Principles of endocrinology. In: Jameson JL, ed. Harrison’s Endocrinology. New York, NY: McGraw-Hill; 2006, Figure 23-1, p. 412.)
Actively resorbing osteoclasts are firmly attached to bone by a zone of membrane that is relatively devoid of subcellular particles (the “sealing” zone). The sealing zone surrounds an area of highly convoluted membrane, the “ruffled border,” where resorption occurs.
Modeling produces growth of the skeleton and changes in bone shape. Linear growth during childhood and adolescence occurs by endochondral bone growth, in which a template of cartilage at the end plates of long bones is replaced by bone formation. The width of the bones increases by periosteal apposition and during childhood, this is accompanied by endosteal resorption to maintain the medullary cavity. The endosteal (or inner) surface is in contact with the marrow; thus, endosteal resorption results in a concomitant enlargement of the marrow cavity. In general, the first bone formed from mesenchyme in early development as well as bone formed during rapid repair has a relatively disorganized pattern of collagen fibers and is termed “woven” bone. Subsequent bone is laid down in an orderly fashion with successive layers of well-organized collagen, and is termed “lamellar bone.”
During puberty and early adult life, endosteal apposition and trabecular thickening provide maximum skeletal mass and strength (peak bone mass). Locally and systemically produced factors and mechanical forces influence these processes. The adult skeleton continues to undergo remodeling throughout life; approximately 15% of the mature skeleton is replaced each year to maintain mineral homeostasis, to repair damaged bone, and to respond to changes in skeletal stress.
Bone remodeling occurs most often in skeletal sites rich in cancellous (trabecular) bone. Trabecular bone is found inside the long bones, in the inner portions of the pelvis and other large flat bones, and throughout the bodies of the vertebrae, where it provides important mechanical support to the spine. Trabecular or cancellous bone constitutes only 20% of the skeleton, but it has an enormous surface area and is particularly active metabolically.
A second form of bone, termed cortical bone, is less metabolically active but provides great strength and integrity to the skeleton. Cortical bone comprises 80% of the skeleton, and constitutes the outer part of all skeletal structures. Cortical bone is dense and compact, and the lamellae may be extensive (circumferential) or tightly packed in concentric circles in osteons. Cortical bone primarily provides mechanical strength and protection, but with severe or prolonged mineral deficits it can participate in metabolic responses.
Bone remodeling is a process that takes place in a cyclic fashion at specific sites or skeletal lacunae. Each remodeling cycle lasts about 90 to 120 days. Bone turnover begins with activation of surface osteoblasts and ostecytes. The signals for initiation of remodeling include changes in the serum concentration of a number of systemic factors, such as PTH, thyroxine, growth hormone, and estrogen. Cytokines and growth factors (eg, IL-1, IL-6, IL-11, insulin-like growth factor I, and TGF-β) trigger the bone-synthesizing activity of osteoblasts.
Resorption—Once cells of the osteoblast lineage are activated, these cells undergo shape changes and secrete collagenase and other enzymes that lyse proteins on the bone surface; they also express a factor that is termed osteoclast differentiating factor or RANK ligand (RANKL). RANKL interacts with a receptor on osteoclast precursors that is identical to the receptor involved in interaction of T cells and dendritic cells called RANK. RANKL stimulates activation, migration, differentiation, and fusion of hematopoietic cells of the osteoclast lineage to begin the process of resorption.5
Osteoclasts express a potent ATPase and carbonic anhydrase, and secrete protons and proteases that dissolve the mineral matrix. Osteoclasts also have acid phosphatase anchored to their cell membranes. During remodeling, multinucleated osteoclasts remove mineral and matrix but stop at a predetermined depth on the trabecular surface or within cortical bone. What stops this process is not clear, but high local concentrations of calcium or substances released from the matrix may be involved.
Reversal—After osteoclastic resorption is completed, a reversal phase occurs, in which mononuclear cells, possibly of monocyte/macrophage lineage, appear on the bone surface. These cells prepare the surface for new osteoblasts to begin bone formation. A layer of glycoprotein-rich material is laid down on the resorbed surface, the so-called “cement line,” to which the new osteoblasts can adhere.
Formation—The formation phase follows, with successive waves of osteoblasts laying down bone until the resorbed bone is completely replaced and a new bone structural unit is fully formed. When this phase is complete, the surface is covered with flattened lining cells, and a prolonged resting period with little cellular activity on the bone surface ensues until a new remodeling cycle begins. The major structural protein in bone matrix is type 1 collagen, which is synthesized by the osteoblasts. Type 1 collagen is the most abundant protein in bone, and is assembled as a complex structure containing two α-1 chains and one α-2 chain. The α-1 and α-2 chains are the products of distinct genes, and after synthesis many of the proline and lysine residues in the collagen chains are hydroxylated to hydroxyproline and hydroxylysine, respectively. Then, one α-2 and two α-1 chains intertwine to form a helical structure known as procollagen, followed by cleavage of their amino-terminal and carboxy-terminal peptides to form tropocollagen.
PTH normally increases the number and the activity of osteoclasts, through at least two mechanisms: upregulation of a factor that is termed RANK ligand (previously known as osteoclast differentiating factor, ODF). RANKL interacts with a receptor on osteoclast precursors that is identical to the receptor involved in interaction of T cells and dendritic cells called receptor activator of NFkB (RANK). RANKL stimulates activation, migration, differentiation, and fusion of hematopoietic cells of the osteoclast lineage to begin the process of resorption. RANKL can also bind a protein that is produced by osteoblasts and other marrow cells called osteoprotegerin (OPG) or osteoclastogenesis inhibitory factor. OPG acts as a “decoy receptor” for RANKL and thereby inhibits osteoclast recruitment and activation. Both PTH and calcitriol act in concert to increase bone resorption and elevate plasma calcium concentration. However, both hormones also have long-term effects that cause inhibition of bone resorption and/or stimulation of bone formation, resulting in a net increase in bone mass. Calcitonin has a direct and acute inhibitory effect on osteoclastic bone resorption and may also decrease the formation of osteoclasts but has no major effect on bone formation.
Glucocorticoids have complex and multiple effects on bone and calcium homeostasis. When used in pharmacologic doses glucocorticoids inhibit both bone formation and resorption. The inhibition of bone formation is probably the major mechanism responsible for the osteopenia associated with glucocorticoid excess.5 Glucocorticoids reduce osteoblast number by decreasing osteoblast formation from precursors in favor of adipocyte generation, inhibit osteoblast activity, and stimulate apoptosis of mature osteoblasts. Glucocorticoids stimulate osteocyte apoptosis and, thus, compromise the ability of bone to respond to stress—leading to decreased bone strength. Conversely, glucocorticoids stimulate expression of RANKL and decrease expression of OPG, thereby extending the life of mature osteoclasts. These antiapoptotic effects would predict increased rather than decreased degradation activity. However, accelerated bone resorption does not appear to contribute to the osteoporosis that occurs with prolonged glucocorticoid therapy; this tempered effect likely arises because glucocorticoids also interfere with osteoclast function.5 Glucocorticoids also negatively impact bone mineralization by impairing intestinal and renal calcium absorption.
Thyroid hormones cause a marked increase in bone turnover, and osteopenia is common in patients with hyperthyroidism, particularly those over the age of 50 years. This effect is likely as a result of a direct stimulatory effect on osteoclastic bone resorption. Insulin hasdirect and indirect effects on both cartilage and bone formation. Some patients with long-standing type 1 diabetes mellitus may develop osteopenia. The effect of insulin is thought to be mediated through increased production of IGF-1 and calcitriol. IGF-1 stimulates protein synthesis and sulfation in cartilage, and both insulin and IGF-1 stimulate bone collagen synthesis. The effects of insulin and IGF-1 on bone are closely related and not additive, suggesting a similar mechanism of action. Growth hormone also stimulates both cartilage and bone formation. This effect is probably mediated through increased production of IGF-1.
Sex steroids accelerate skeletal growth and bone maturation, and contribute to bone accumulation and are antiresorptive. These effects may be direct or conferred through interaction with the GH system or mechanical loading. Estrogens alone and estrogens in combination with testosterone increase bone mineral density to a greater extent than testosterone alone. Moreover, bone formation is reduced in males who are unable to produce estrogens due to genetic defects in aromatase, suggesting that much of the effect of androgen action on bone is mediated through estrogens.6 The osteoclast is the target of antiresorptive effects of estrogen; the androgen site is not yet defined.7 The estrogen receptor-α is responsible for at least some of the estrogen effects as highlighted by the abnormal bone histology described in a male with genetic mutations that led to a loss of estrogen receptor-α function.8 Direct effects of androgens, however, are also important and are mediated through the androgen receptor which is expressed in pluripotent mesenchymal stem cells, osteoblasts, osteoclasts, and osteocytes. Osteocyte/osteoblast-specific androgen receptor knock-out mice highlight the role of androgens in maintaining trabecular bone.9 Androgens, through accrual of lean mass, also target periosteal bone apposition.
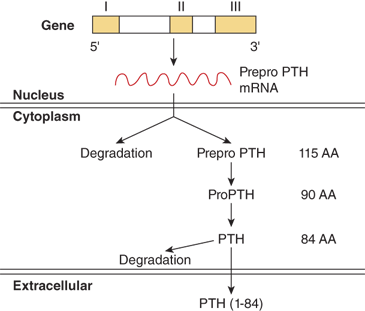
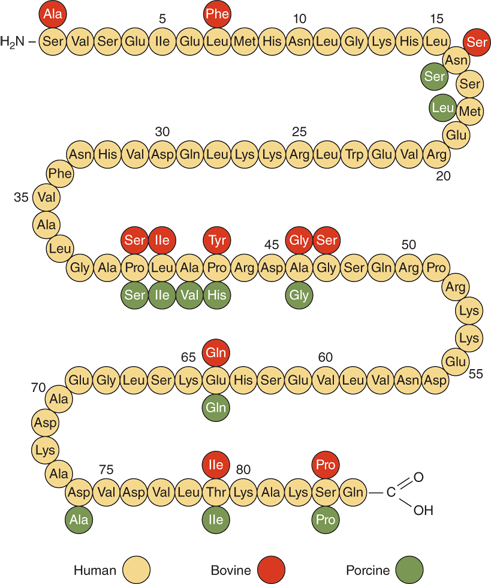
PTH is synthesized and secreted by chief cells, the major cells of the parathyroid glands. PTH is produced by a two-step proteolytic conversion of the primary gene product, prepro PTH (105 amino acids), with cleavage of the 25-amion acid leader sequence to yield proPTH (90 amino acids) and then the mature PTH molecule (Figure 7-6), a single chain polypeptide comprised of 84 amino acids with a molecular weight of 9500 daltons (9.5 kDa) (Figure 7-7). Biological activity, as defined by hypercalcemic potency, resides within the first 34 amino acids at the amino terminus, which shows the greatest degree of sequence homology among different species and with the related molecule, PTHrP. The first two amino acids are essential for activation of the receptor, and the region comprised of amino acids 3 to 34 contains the structural determinants necessary for receptor binding.
PTH synthesis and secretion from the parathyroid glands occurs at a rate that is inversely proportional to the serum ionized calcium concentration. PTH release is pulsatile and temporally coupled with the plasma concentrations of calcium and phosphate. In addition, a circadian periodicity in plasma PTH concentrations exists with a characteristic nocturnal increase. PTH secretion is also inhibited by calcitriol and FGF23, and is increased by extracellular phosphate.
Extracellular calcium, and to a lesser extent other divalent cations, regulate PTH synthesis and secretion through interaction with specific calcium-sensing receptors (CaSRs) that are expressed on the surface of parathyroid cells. These receptors are also present on many other cells throughout the body, including the calcitonin-secreting C cells of the thyroid and renal tubular cells. The CaSR is a G protein–coupled receptor (GPCR) and consists of a single polypeptide chain that spans the plasma membrane seven times (ie, heptahelical), forming three extracellular and three or four intracellular loops and a cytoplasmic carboxyl-terminal tail. CaSRs interact with heterotrimeric (αβγ) G proteins that belong to the Gq/11 and Gi/o families to regulate activityof signal effector molecules (eg, adenylyl cyclase, phospholipase C β, potassium channels) that are localized to the inner surface of the plasma membrane.
Binding of extracellular calcium to the CaSR activates the receptor and facilitates its interaction and activation of G proteins that stimulate phospholipase C β activity (Gq and G11) and inhibit adenylyl cyclase (Gi). Activation of phospholipase C leads to the generation of the second messengers inositol 1,4,5-trisphosphate and diacylglycerol, which increase levels of cytosolic calcium via release from intracellular stores and stimulate protein kinase C activity, respectively. These second messenger systems mediate the parathyroid cell’s responses to elevated concentrations of extracellular calcium, which include inhibition of PTH release, suppression of PTH gene expression, and accelerated degradation of intracellular PTH.
A decrease in extracellular calcium rapidly increases the secretion of PTH from a pool of stored intracellular hormone that is sufficient to sustain PTH secretion for only 60 to 90 minutes. Under conditions of protracted hypocalcemia the parathyroid cell relies upon two mechanisms to increase PTH availability: first, inhibition of intracellular hormone degradation and second, increased levels of PTH mRNA (with hyperplasia if needed). A basal, nonsuppressible PTH secretion that is independent of extracellular calcium also exists and is proportional to the mass of chief cells. As a result, basal PTH secretion is markedly increased by parathyroid gland hyperplasia.
The extracellular magnesium concentration also seems to have a role in the regulation of PTH secretion. Severe magnesium deficiency results in decreased PTH secretion as well as refractoriness to PTH action in target cells and can cause hypocalcemia. Conversely, elevated serum levels of magnesium can directly inhibit PTH secretion via direct interaction with CaSRs.
Once secreted into the circulation, PTH has a short half-life, probably on the order of less than 5 minutes. It is taken up by hepatic Kupffer cells, where cathepsin enzymes cleave the region between amino acids 33 and 43, thereby generating amino- and carboxy-terminal fragments. In addition, fragments consisting of PTH 7-84 are also generated, which can inhibit PTH 1-84 action. Carboxyl region fragments are released back into circulation, whereas the amino region portions are probably further degraded so that they are not released as bioactive entities. The kidney is also known to have PTH cleaving enzymes, but apparently neither carboxyl nor amino region fragments are released back into the circulation from the kidney.
The primary function of PTH is to increase the extracellular fluid calcium (1) directly, by stimulating the rates of renal calcium reabsorption and bone resorption and (2) indirectly, by increasing the rate of intestinal calcium absorption via increased formation of calcitriol in the kidney. These PTH effects are mediated by specialized target cells present in bone and kidney that express specific plasma membrane G protein–coupled receptors. The classical PTH receptor is an approximately 75-kD glycoprotein that is often referred to as the PTH/PTHrP or type 1 PTH receptor (PTH1-r). The PTH1-r is expressed on bone and kidney cells and binds both PTH and PTHrP, a factor made by diverse tumors that cause humorally mediated hypercalcemia, with equivalent affinity. The PTH1-r is most abundantly expressed in the physiological target tissues for PTH action (ie, kidney and bone), but it is also found in a wide variety of fetal and adult tissues where it appears to mediate the paracrine/autocrine signaling pathways of PTHrP rather than the endocrine actions of PTH. By contrast, a second PTH receptor, termed the type 2 receptor protein, interacts with PTH but not PTHrP, and has a very restricted tissue distribution that does not include classical PTH target tissues (ie, bone and kidney). It is more likely that the hypothalamic peptide termed tuberoinfundibular peptide of 39 residues (TIP39) is the physiological ligand for the type 2 PTH receptor,10 but the biological functions mediated by this receptor remain largely unknown.
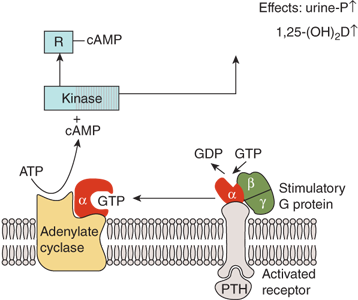
The PTH1-r couples to G proteins that stimulate adenylyl cyclase (Gs) (Figure 7-8) and phospholipase C (Gq/11), to activate protein kinase A and protein kinase C signaling pathways, respectively. Thus, receptor activation leads to rapid generation of the second messengers cAMP, inositol 1,4,5-trisphosphate and diacylglycerol, and cytosolic calcium.
Figure 7-8
Mechanism of action of PTH in the kidney. The activation of adenylate cyclase is mediated by the Gs-protein which consists of α-, β-, and γ-subunits. The α-subunit contains the guanine nucleotide-binding site that, in the nonactivated state, is occupied by GDP. Binding of PTH to the receptor induces a change in the conformation of the α-subunit leading to an exchange of bound GDP for GTP. The α-GTP complex then dissociates from the rest of the Gs-protein and activates adenylate cyclase. Cyclic AMP is produced and binds to the regulatory subunit (R) of cAMP-dependent protein kinase, liberating the catalytic subunit of the enzyme which subsequently phosphorylates and activates enzymes involved in tubular phosphate transport and 1-α-hydroxylation of 25(OH)D.
The renal handling of phosphate appears to be the main determinant of plasma phosphate concentration. PTH inhibits phosphate reabsorption, thereby resulting in phosphaturia and, in turn, lower plasma phosphate concentrations. PTH mediates this effect by controlling the concentration of Npt2a and Npt2c cotransporters on the brush border membrane located at the apical surface of proximal tubular cells. In hypoparathyroid states TmP/GFR is set at a higher level resulting in increased phosphate resorption and increased plasma phosphate concentration.
PTH-enhanced reabsorption of calcium in distal tubules is mediated directly (and indirectly via calcitriol) by increased expression or activity of the transient receptor potential cation channel 5 (TRPV5) (see Figure 7-3). In hyperparathyroidism the opposing forces of increased filtered load because of hypercalcemia and increased tubular reabsorption due directly to PTH determine the urinary excretion of calcium. PTH also has a minor effect on tubular transport of bicarbonate and amino acids so that aminoaciduria and a rise in pH and bicarbonate content of the urine can be present in hyperparathyroid states. PTH stimulates the rate-limiting enzyme in vitamin D activation, 1-α-hydroxylase in the kidney, (see section “Vitamin D” stated later on). Thus, PTH indirectly increases intestinal calcium and phosphate absorption by stimulating calcitriol production in the proximal tubule.
Osteoblasts possess PTH receptors, whereas osteocytes and mature osteoclasts do not appear to bind PTH. The available data indicate that PTH acts primarily on mature osteoblasts to enhance their function, their lifespan, or both, and it may also enhance the differentiation of cells in the osteoblastic lineage. These findings indicate that the PTH stimulation of osteoclastic bone resorption is mediated through osteoblasts. One of the most important consequences of PTH action is mediated via increased expression of RANK ligand (RANKL) on the cell surface (described in section “Action in the Kidney” stated earlier). PTH also decreases production of osteoprotegerin (OPG), a soluble decoy receptor that can bind RANKL and block its interaction with RANK.11 By increasing production of RANKL and decreasing production of the natural antagonist OPG, PTH increases osteolastic bone resorption. PTH and calcitriol act in concert to elevate the plasma calcium by stimulating bone resorption.
PTH also increases bone formation in a manner that appears to require IGF-1. PTH stimulates osteoblasts to synthesize and secrete IGF-1 into the local microenvironment, where it behaves as an autocrine or paracrine factor to stimulate osteoblasts and osteoblast precursors.12
Immunometric “sandwich” assays have largely replaced early radioimmunoassays for PTH that detected principally biologically inactive mid- and carboxy-terminal PTH fragments. Current immunometric assays for intact PTH not only detect the intact hormone, but also additional PTH fragments that are truncated at the amino terminus and consist principally of PTH (7-84). More recent “second-generation” immunometric PTH assays use a detection antibody that recognizes antigenic determinants at the extreme amino-terminal (1-4) end of the PTH molecule, making the assay specific for biologically active “whole” PTH (1-84). These whole molecule assays will not detect PTH (7-84) fragments, which are directly released from the parathyroid gland in humans and appear to exert a hypocalcemic effect by inhibiting the calcemic effect of PTH (1-84) and its stimulatory effect on bone turnover. Because the half-life of 1-84 PTH is much shorter than that for the 7-84 PTH fragment, the development of this assay has been most useful for monitoring PTH levels in patients with renal failure where PTH 7-84 fragments accumulate to excess. By contrast, in general clinical practice immunoassays for whole PTH 1-84 appear to offer little advantage over assays for intact PTH.
PTHrP was originally discovered as the cause of humorally mediated hypercalcemia of malignancy. PTHrP interacts with shared PTH/PTHrP (PTH1-r) receptors in the kidney and bone through its amino terminus, which is homologous with the amino terminus of PTH. The PTH and PTHrP genes have presumably evolved from a common ancestor through an ancient gene duplication event. Overall, PTH and PTHrP share significant amino acid homology only at the amino terminus, where 8 of the first 13 amino acids are identical. Remarkably, even in the primary receptor binding domain (amino acids 18-34), PTH and PTHrP do not have recognizable primary sequence homologies. PTH and PTHrP bind to the type 1 PTH receptor with equivalent affinity, which no doubt explains why biological actions that are mediated via the amino terminus of the two molecules appear to be very close in both range of biological activities and potency.
Compared to PTH, PTHrP is considerably longer, with variants containing either 139, 141, or 173 amino acids. PTHrP plays a significant role in regulating skeletal development and fetal calcium homeostasis, and is widely expressed in adult tissues including cartilage, bone, smooth muscle, placenta, central nervous system (CNS), and epithelial tissues such as breast, hair follicle, intestine, and tooth enamel. In addition, PTHrP has a nuclear localization signal in its midportion that directs the hormone to the nucleus, where it appears to directly regulate transcription of several target genes.
The observation by Palm in 1890 that rickets could be treated by exposure to sunlight marked the beginning of a scientific quest that led to the discovery of vitamin D by Windaus and coworkers in 1932. The appreciation that parent vitamin D must undergo two activation steps to be converted to the wholly active form calcitriol, along with the elucidation of a molecular mechanism of action analogous to that of classic steroid hormones, led to the recognition that parent vitamin D is a hormone precursor rather than merely an essential nutritional substance (ie, vitamin). The distribution of vitamin D receptors in many tissues in addition to the intestine, the skeletal system, and the kidney implies that the hormone has biological functions beyond its classical role in mineral homeostasis.
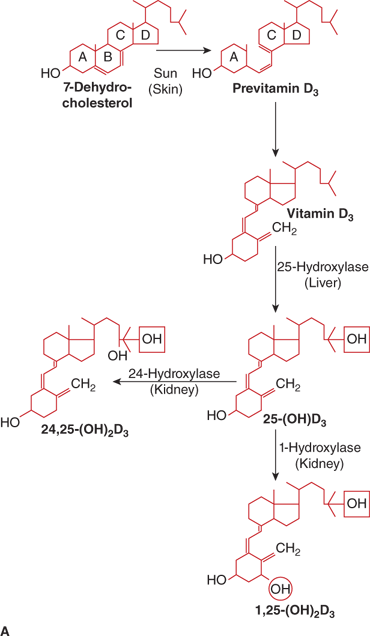
The calciferols (vitamin D) constitute a family of fat-soluble biologically active secosteroids (Figure 7-9). The term vitamin D refers to both vitamin D2 (ergocalciferol) that originates in plants and to vitamin D3 (cholecalciferol) produced in the body. Both vitamin D2 and vitamin D3 are used to fortify foods. Vitamin D2 and vitamin D3 have generally been considered to be of near-equal potency in humans,13 but some studies suggest that vitamin D3 may be slightly (two- to threefold) more potent than vitamin D2 in humans in maintaining serum levels of 25-hydroxyvitamin D [25(OH)D]14 as vitamin D3 and its 25-hydroxylated metabolite may bind vitamin D–binding protein (DBP) with greater affinity than vitamin D2 (see below).14 Vitamin D3 is produced in the skin via opening of the B-ring of 7-dehydrocholesterol. During exposure to sunlight, 7-dehydrocholesterol absorbs solar radiation with energies between 290 and 320 nm (UVB ultraviolet light), and is photocatalytically transformed into previtamin D3. Over the next several hours previtamin D3 isomerizes to vitamin D3 by a temperature-dependent process. This process permits the skin to continue the synthesis of vitamin D3 for up to 3 days after a single exposure to sunlight. Once vitamin D3 is formed, it is translocated to the plasma via binding to DBP. Excessive sunlight exposure is unlikely to cause vitamin D intoxication, as previtamin D can be degraded by further UVB radiation to the inert photoproducts lumisterol and tachysterol and vitamin D3 can be photoisomerized to suprasterol and other biologically inactive products.
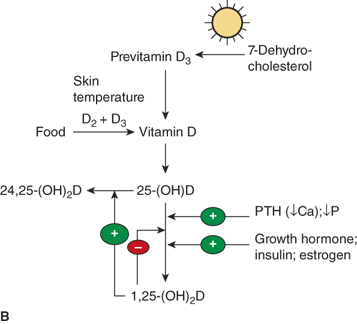
Figure 7-9
(A) Pathway of vitamin D metabolism. The rate-limiting step in the activation of vitamin D is the 1-α-hydroxylation of 25(OH)D3 to calcitriol and is directly stimulated by PTH and low serum phosphate concentration. (B) Regulation of vitamin D metabolism. PTH is the main stimulator of calcitriol synthesis. The effect of ionized calcium is indirect and mediated by PTH. Calcitriol inhibits its own synthesis and stimulates the production of 24,25(OH)2D3, an inactive vitamin D metabolite.
For most individuals, daily requirements for vitamin D can be satisfied by exposure of hands, face, and arms to sunlight for 20 to 30 minutes three times per week. Many factors can influence the efficiency of cutaneous production of vitamin D. Sunscreens, even those with a sunlight protection factor rating as low as 8 (SPF 8) can markedly reduce, by as much as 97%, the cutaneous production of vitamin D. Moreover, chronic use can result in vitamin D deficiency. The melanin in skin pigment absorbs UVB photons and thereby behaves as a natural sunscreen, such that dark-skinned people will require greater exposure (5- to 10-fold) than light-skinned people for cutaneous synthesis of equivalent amounts of vitamin D. Similarly, street clothing can decrease the amount of cutaneous vitamin D synthesized in response to UVB radiation. Latitude, time of day, and season can also affect the efficiency of solar irradiation on cutaneous synthesis of vitamin D. Very little cutaneous vitamin D is formed during the winter months (October through March in the northern latitudes and April through September in the southern latitudes) owing to changes in the angle of the winter sun that reduce the amount of UVB irradiation that reaches the earth’s surface. In addition, sunlight exposure is further limited by increased clothing worn and decreased time spent outdoors during winter. During sunlit hours, vitamin D3 production is maximal at midday and small quantities are still being formed between 08:00 and 09:00, and between 16:00 and 17:00 during the summer. Full-body irradiation can produce as much as 10,000 to 20,000 IU of cholecalciferol, and for most individuals, the cutaneous production of vitamin D during the spring, summer, and fall is in excess of daily requirements. During the winter months, fat-stored cholecalciferol (3000-5000 IU/day) is used to provide more than 80% of daily needs. Finally, aging also reduces the ability of skin to synthesize vitamin D.
Humans obtain calciferols either endogenously through metabolism of precursors in the skin, or exogenously as a dietary component or dietary supplement. For practical purposes, the metabolism and actions of vitamin D3 and vitamin D2 are similar in humans, although slight distinctions in the processing of these two steroids have been recently noted. In the temperate zones vitamin D status is mainly determined by exposure to solar irradiation, and among individuals who spend significant time outdoors the serum concentration of 25(OH)D can reflect season, global latitude, and average hours of sunlight exposure.
Vitamin D must undergo two hydroxylation steps to achieve full biological potency (see Figure 7-9). Once vitamin D2 or vitamin D3 enters the circulation it is transported by the DBP to the liver to form 25(OH)D. Although several hepatic cytochrome P450 enzymes can perform this step, the principal 25-hydroxylase is CYP2R1 that is present in the microsomal fractions of the liver cells.69 At normal circulating concentrations, 25(OH)D exerts little if any vitamin D effect due to its relatively low affinity for the vitamin D receptor (VDR) that is present in the cell nucleus. Nevertheless, as 25(OH)D is the principal form of vitamin D in the circulation, the plasma concentration of 25(OH)D is considered the best indicator of a person’s vitamin D status. Available evidence suggests that the 25-hydroxylation step is not tightly regulated, even though a mild degree of product feedback inhibition may occur.
To achieve full potency as a ligand for the VDR, 25(OH)D must undergo a second hydroxylation, at the C1 position, to become the hormonal form, calcitriol. The 1-α-hydroxylase enzyme system is located in the mitochondria of the proximal convoluted and straight tubules of the nephron and appears to be a classical mixed-function cytochrome P450 steroid hydroxylase (CYP27B1). Activity of renal CYP27B1 is tightly regulated, such that production of calcitriol can vary to meet an individual’s unique needs as long as adequate amounts of substrate 25(OH)D are available. PTH activates enzyme activity through a cAMP-mediated pathway. Enzyme activity is also stimulated directly by calcitonin and indirectly by growth hormone and prolactin. Calcitriol, phosphate, FGF23, and calcium inhibit CYP27B1 activity and reduce 1-α-hydroxylation of 25(OH)D. In humans, the circulating concentration of calcitriol is approximately one thousandth that of 25(OH)D.
In the past, the kidney was considered the sole site of calcitriol production. However, the identification of elevated plasma calcitriol concentrations in an anephric patient with sarcoidosis led to the recognition that 1-α-hydroxylation of 25(OH)D could occur in other tissues.15 Subsequently, synthesis of calcitriol was demonstrated in alveolar macrophages and in lymph node homogenates from patients with sarcoidosis. Elevated plasma levels of calcitriol have also been observed in hypercalcemic patients with other granulomatous disorders such as candidiasis, tuberculosis, subcutaneous fat necrosis of the newborn, and in silicon-induced granulomas. In addition, in vitro studies have disclosed that tissues such as placenta, colon, breast, as well as osteoblasts, activated macrophages, and keratinocytes express CYP27B1 activity and are capable of converting 25(OH)D to calcitriol. Local calcitriol production may serve an autocrine role in regulating cell growth and/or other activities as these tissues and cells also express vitamin D receptors. In fact, the role of PTH-independent vitamin D metabolism in immune regulation has received much attention with respect to cancer, infections, and immune disorders.
Both calcitriol and 25(OH)D are catabolized by CYP24 (also known as 24-hydroxylase) in the kidney, which is followed by sequential metabolism, yielding the terminal product calcitroic acid. Calcitriol and 25(OH)D can also be degraded by hepatic and intestinal CYP3A4, which leads to 23- and 24-hydroxylations of vitamin D metabolites (see Figure 7-9). The balance between bioactivation and degradation of calcitriol ensures appropriate biological effects and is tightly controlled in vivo. In normal prepubertal and pubertal children the plasma concentration of (relatively inactive) 24,25-(OH)2D is approximately 50 to 100 times that of calcitriol and 3% to 6% that of 25(OH)D. Another nuclear receptor, the steroid and xenobiotic receptor (SXR; also known as pregnane X receptor [PXR], PAR, and NR1I2) is expressed at high levels in the liver and small intestine, where it acts as a xenobiotic sensor that regulates the expression of CYP3A4 and other enzymes that are involved in xenobiotic metabolism. A variety of drugs (eg, rifampin, phenobarbital, carbamazepine, etc) that bind to SXR can induce expression of CYP3A4 and thereby accelerate the degradation of calcitriol and 25(OH)D.16
Over 99% of vitamin D metabolites circulate bound to plasma proteins. DBP is the principal carrier protein, but albumin also contributes to transport. DBP is an α-globulin of 58,000 D and is synthesized by the liver. It is a major component of plasma (normal concentration 10−5 M) and demonstrates considerable genetic variability with many different isoforms that exhibit variable affinities for vitamin D metabolites. In fact, the highly polymorphic nature of DBP made it an appealing target for studies of genetic diversity as “Gc globulin” long before its role in vitamin D homeostasis was recognized. The DBP binding site for vitamin D metabolites also binds fatty acids; a separate binding site exists for actin (reviewed in Malik et al17).18 The DBP is also the precursor to the immunomodulatory protein, Gc-MAF. Two missense variants of the DBP gene—rs7041 encoding Asp432Glu and rs4588 encoding Thr436Lys—change the amino acid sequence and alter the protein function. These variants are sufficiently common to generate population-wide constitutive differences in vitamin D status, based on assay of the serum metabolite, 25-hydroxyvitamin D (25OHD).
The major vitamin D metabolites are bound to a single high-affinity binding site of DBP with the following order of affinities: 25(OH)D3-26,23-lactone > 25(OH)D = 24,25(OH)2D > 1,25(OH)2D >> vitamin D. DBP is a highly expressed protein with multiple functions, including transport of vitamin D metabolites, control of bone development, binding of fatty acids, sequestration of actin and a range of less-defined roles in modulating immune and inflammatory responses. Unlike other hormone-binding proteins that show nearly complete occupancy of all binding sites, less than 1% of DBP’s binding sites are occupied. Under normal circumstances approximately 85% of total circulating calcitriol is bound to DBP, about 15% to albumin, and about 0.03% is free. Only the free fraction of circulating calcitriol is thought to exert metabolic effects in target cells. The plasma concentrations of DBP and, hence, total calcitriol are increased during pregnancy and after intake of oral contraceptives and estrogen, whereas the level of the free and presumably physiologically active fraction remains essentially unchanged. DBP may decrease with severe illness, nephrosis, liver disease, or surgery—complicating interpretation of 25(OH)D in these settings.18 Polymorphisms in DBP vary by ancestry (African American, Caucasian, Hispanic) and partly explain differences in measured total and free 25(OH)D. Moreover, while total 25(OH)D are not associated with bone mineral density, both free and bioavailable 25(OH)D levels have been associated with BMD in adults after adjustment for age, sex, body mass index, and race.19 At present, data to support a role for DBP in the vitamin D endocrine system beyond that of a transport protein remain scant.
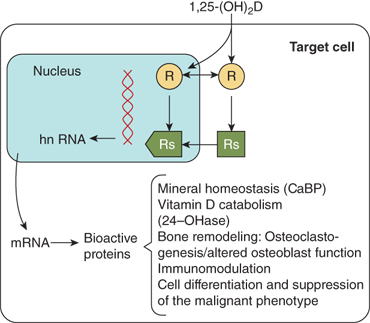
The biological actions of calcitriol are mediated through the vitamin D receptor (VDR), a 50-kDa intracellular nuclear receptor that functions as a transcriptional regulator in a manner similar to that described for classical steroid hormone receptors (eg, glucocorticoid, androgen, and estrogen).20 The VDR is highly expressed in cells of the small intestine, kidney and bone, the major classical vitamin D target tissues, and also in other tissues including testis, ovary, pituitary, parathyroid, skin, breast, muscle, lymphocytes, and some tumor cell lines. The VDR shares a structure that is similar to other steroid hormone receptors, and contains discrete functional domains, including a hormone-binding domain at the carboxy terminus and a DNA-binding domain at the amino terminus characterized by two fingers anchored via Zn2+ atoms to cysteine residues (see Chapter 1). The binding affinity of different vitamin D metabolites is correlated with their biological potencies. Calcitriol is the most potent natural calciferol metabolite, with a binding affinity that is approximately 1000-foldgreater than that of 25(OH)D in most test systems. Calcitriol binds to the hormone-binding domain of the VDR, and the VDR must complex with the retinoic acid X receptor (RXR) to form a heterodimeric complex. The DNA-binding domain of the VDR is then able to bind to a vitamin D response element (VDRE) in target genes, thereby regulating gene transcription to increase or decrease the synthesis of specialized proteins mediating the biological effect of the hormone (Figure 7-10). Many of the target genes are known (eg, intestinal calcium binding proteins, osteocalcin, TRPV5, VDR, CYP27B1 and CYP24, osteopontin, and calmodulin [Table 7-2]). Calcitriol also has some cellular effects that are not mediated by a genomic pathway.
The most important physiologic action of calcitriol is stimulation of active calcium transport across the duodenum from lumen to bloodstream. Vitamin D compounds are believed to increase transcellular calcium absorption by inducing expression of the Ca2+-selective channels TRPV5 and TRPV6 on the luminal membrane, cytosolic calcium-binding proteins (eg, calbindin D9k), and the calcium transporters PMCA1b and NCX1 on the basolateral membrane of enterocytes in the duodenum and upper jejunum. The intestinal absorption of phosphate occurs principally in the jejunum and ileum, and is less dependent on calcitriol than is the case for calcium. Hence, in vitamin D deficiency, the reduction of phosphate absorption is proportionally much less than the reduction of calcium absorption. Specifically, adults who are vitamin D replete can absorb approximately 30% of dietary calcium and 70% to 80% of dietary phosphate, whereas adults who are vitamin D deficient can absorb no more than 10% to 15% of dietary calcium and 60% of dietary phosphate.
Calcitriol is essential for normal bone growth and mineralization. The effect on bone mineralization is probably indirect by providing minerals for incorporation into bone matrix through increased intestinal absorption of calcium. Calcitriol inhibits proliferation and collagen synthesis in fetal bone and in fetal osteoblasts, whereas in osteoblast-like cells from adult humans calcitriol stimulates collagen synthesis. Differing effects on alkaline phosphatase have been reported in several systems; however, there is general agreement that in rapidly growing osteoblast-like cells alkaline phosphatase levels are low and that they rise in response to calcitriol (see Table 7-2).
Although vitamin D is essential for bone mineralization, calcitriol also reduces deposition of minerals into bone and stimulates bone resorption. Although these direct effects on the skeleton appear counter intuitive, they likely reflect the important role of calcitriol to maintain physiological plasma levels of calcium and phosphorus. In vivo and in organ culture, calcitriol is a potent activator of osteoclasts. Calcitriol induces monocyte stem cells in the bone marrow to differentiate to osteoclasts. Once osteoclasts have matured they no longer respond directly to calcitriol and do not contain VDRs. Osteoclastic activity is stimulated indirectly by calcitriol through its action on osteoblasts and osteocytes, which produce a variety of cytokines, including RANKL.
Probably the most important effect of calcitriol on the kidney is inhibition of its own synthesis through suppression of CYP27B1 and the concomitant enhancement of its degradation through induction of CYP24. These actions are both direct, through genomic effects, and indirect via induction of FGF23 (see in section “Regulation” stated later).21 Calcium absorption in the distal tubule is also regulated by calcitriol through its ability to induce expression of the TRPV5 calcium channel, calbindin-D28k, and calbindin-D9k, and the basolateral membrane calcium transporters NCX1 and PMCA1b.
The conversion of 25(OH)D to the active hormonal form calcitriol is highly regulated. The factors include PTH, calcium, phosphate, calcitriol itself, FGF23, and various other hormones (Figure 7-11). PTH is the main trophic hormone for 1-α-hydroxylation, and elevated calcitriol levels are found in both primary and secondary hyperparathyroidism. Additionally, calcitriol induces expression of the VDR, and therefore amplifies its own action through a feed forward mechanism. Calcitriol inhibits its own synthesis via inhibition of CYP27B1 in the kidney, whereas there seems to be little effect of calcitriol on expression of CYP27B1, and hence production of calcitriol, in other tissues such as alveolar macrophages. The regulatory effects of calcium and phosphate are both likely indirect and mediated by PTH and FGF23, respectively. According to this concept, hypocalcemia stimulates PTH secretion, which, in turn, stimulates calcitriol production, leading to (1) increased circulating calcium, which, inhibits PTH secretion and (2) increased phosphate absorption, thereby offsetting PTH-mediated phosphaturia.
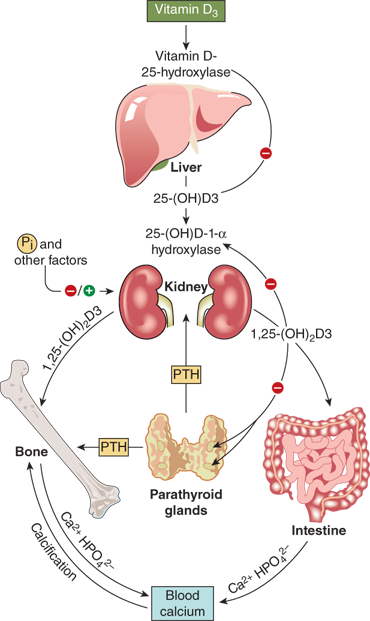
Dietary phosphorus seems to have more of a long-term regulatory effect. Phosphate restriction and hypophosphatemia produce an increase in calcitriol plasma concentration within several days, whereas a high intake of phosphate decreases the level of calcitriol. FGF23 (and possibly additional phosphatonins) appears to mediate these effects.
Several other endocrine factors (GH, IGF-1) have direct or indirect effects on renal 1-α-hydroxylase activity, although the exact relationships and their clinical significance remain to be clarified (see Figure 7-11). Growth hormone and IGF-1 may also be involved in mediating the increase in serum calcitriol concentration induced by restriction of dietary phosphorus. Newly diagnosed children with insulin-dependent diabetes have moderately reduced plasma concentrations of calcitriol which increase to normal within 3 weeks of insulin treatment. Decreased circulating levels of calcitriol have been described in hyperthyroidism, and, in contrast, elevated levels in hypothyroidism. Estrogen treatment in relatively high doses leads to a modest increase in the serum concentration of calcitriol. This effect is attributed to a concomitant rise in DBP, resulting in a selective increase of the protein-bound fraction, and essentially unchanged level of biologically active free calcitriol fraction. Pubertal girls with anorexia nervosa have low serum concentrations of calcitriol despite normal concentrations of DBP, which results in low levels of the unbound free fraction.
The renal 24-hydroxylase CYP24 responds to many of the same modulators as the 1-α-hydroxylase, but in the opposite direction (see Figure 7-11). Accordingly, calcitriol inhibits renal production of calcitriol via suppression of CYP27B1, and enhances the formation of 24,25-(OH)2D and 1,24,25 (OH)3D via induction of renal CYP24. The induction of 24-hydroxylase by calcitriol has also been found in other tissues that express calcitriol receptors. An assessment of the 24,25-(OH)2D synthesizing capacity has therefore been extensively used to indicate receptor-mediated responsiveness of a given tissue to calcitriol.
During the last few years a series of discoveries has suggested that calcitriol may influence the proliferation and differentiation of both normal and malignant tissues, and that the hormone may also have immunoregulatory properties. Most cells in the body express the VDR, although the exact physiological function of calcitriol in many nonclassical target tissues remains obscure (Table 7-3).
Although calciferols and 1,25-(OH)2D can be measured in the plasma, the best indication of vitamin D status is afforded by measurement of 25(OH)D as 25(OH)D is produced constitutively from dietary and cutaneous calciferols, and thus represents substrate availability, and 25(OH)D is the most abundant form of vitamin D in the circulation. The relatively long serum half-life (~3 weeks) of circulating 25(OH)D also allows blood samples that are obtained at any time during the day to provide information about steady-state levels. By contrast, parent vitamin D, in the form of cholecalciferol or ergocalciferol, has a short half-life (~24 hours) so that serum levels depend on recent sunlight exposure and vitamin D ingestion. Moreover, measurement of calciferols is challenging owing to the lipophilic nature of the molecule, and clinical assays are not available. Measurement of 1,25(OH)2D, the activated form of vitamin D, is also not a reliable indicator of vitamin D status. As described previously, formation of 1,25(OH)2D is tightly regulated by the action of PTH, and in vitamin D deficient states excess production of PTH (secondary hyperparathyroidism) stimulates the kidneys to produce even more 1,25(OH)2D, such that levels can appear to be normal or even paradoxically elevated. The 25(OH)D metabolite, which reflects total body bioavailability of the prohormone, is therefore the commonly accepted measure of vitamin D status.
Because most 25(OH)D in the circulation is protein bound, chromatographic assays require an extraction step to release 25(OH)D from its binding proteins, which can be subject to variable coprecipitation. On the other hand, nonextracted immunoassay methods may be susceptible to matrix effects, particularly owing to the lipophilic nature of 25(OH)D. In the context of the low (nanomolar) levels of vitamin D metabolites in serum, these factors have made the routine measurement of 25(OH)D an analytical challenge.
Many reference laboratories now use HPLC or liquid chromatography with tandem mass spectrometry (LC-MS/MS) techniques to measure plasma levels of 25(OH)D2 and 25(OH)D3; this approach is considered the gold standard. Many hospital clinical laboratories still use immunoassays, though. Because not all currently available immunoassays detect 25(OH)D2 and 25(OH)D3 with similar sensitivity, the 25(OH)D could be seriously underestimated using the immunoassay in individuals supplementing with ergocalciferol, and, dangerous recommendations for additional supplementation could be erroneously made. Regardless of the methodology used to measure 25(OH)D, standardization of results between methods and laboratories remains an important challenge, and not all hospital laboratories participate in national quality control programs. The Diasorin RIA shows good correlation with HPLC in experienced hands but may not perform well in a laboratory that has not validated the method.
The plasma 25(OH)D concentration reflects the vitamin D status of an individual and is primarily determined by sunlight exposure and the dietary supply of parent vitamin D. Seasonal variation must be taken into account when subjects experience significant sunlight exposure, with the highest concentrations observed in late summer and lowest in late winter. Recent studies now highlight the significant controversy surrounding the definition of the normal or optimal serum levels of 25(OH)D. Traditionally, serum 25(OH)D levels below 5 to 10 ng/mL were recognized to lead to rickets or osteomalacia and serum levels above 10 to 12 ng/mL were considered normal or adequate. However, observational and experimental studies have shown that serum PTH levels are stabilized and intestinal calcium absorption is optimized when serum 25(OH)D levels are greater than 31 ng/mL (78 nm/L).22 In addition, several observational studies have demonstrated incomplete mineralization of bone in subjects who had serum concentrations of 25(OH)D between 20 and 30 ng/mL. Hence, a revised classification of vitamin D status has been proposed by both the American Society for Bone and Mineral Research as well as the Endocrine Society that defines sufficient levels of serum 25(OH)D as 30 to 100 ng/mL (80-250 nmol/L), with levels between 20 and 29 ng/mL (25 and 75 nmol/L) as vitamin D insufficiency, and levels of 25(OH)D that are below 20 ng/mL (25 nmol/L) as deficient.23 This new classification attempts to correct previous schemes that defined the normal range for serum 25(OH)D concentrations using statistical analyses (ie, mean ± 2 SDs) of values obtained in populations that undoubtedly included many individuals who were vitamin D insufficient or deficient. By contrast, the Institute of Medicine (IOM) reviewed similar studies and proposed that a serum concentration of 25(OH)D that is greater than 20 ng/mL represents a sufficient level24; The IOM guidelines were recently adopted by the American Academy of Pediatrics.25 One proposed compromise between these two different standards is to view a serum level of 25(OH)D greater than 20 ng/mLas sufficient, and a serum level of 25(OH)D greater than 30 ng/mL as optimal.
In normal children the plasma concentration of calcitriol ranges between 25 and 85 pg/mL (60-120 pmol/L), with the higher values seen in infancy and adolescence. These differences probably reflect increased intestinal mineral absorption needed during periods of rapid growth. The plasma concentration of calcitriol is not affected by sunlight exposure, and does not increase after administration of vitamin D unless the serum level of PTH is elevated (eg, primary or secondary hyperparathyroidism). The other two dihydroxylated metabolites 24,25(OH)2D and 25,26(OH)2D show seasonal variation and their plasma concentrations are positively correlated to 25(OH)D. In normal children the plasma concentration of 24,25(OH)2D ranges between 3% and 6% of the 25(OH)D level, whereas the concentration of 25,26(OH)2D is about 1% of the 25(OH)D level.
The recent characterization of specific disorders of phosphate metabolism has revealed important roles for phosphatonins, a family of hormones that regulate phosphate homeostasis. The best characterized phosphatonin is FGF23, a member of the FGF superfamily, which is comprised of 22 members that are grouped into seven subfamilies. The paracrine FGFs require the presence of heparin sulfate glycosaminoglycans to allow signal transduction, resulting in a variety of effects including embryonic development, tumor growth, angiogenesis, and wound healing. FGF23 is a 32-kDa protein expressed in osteocytes and osteoblasts, and belongs to the FGF19 family of secreted FGFs, which also includes FGF19 and FGF21. The secreted FGFs can function as endocrine hormones because they have reduced affinity for heparin sulfate, which enables them to escape irreversible association with the extracellular matrix. FGF23 and other members of the FGF19family have reduced affinity for the FGFRs, and binding is dependent on the presence of the coreceptor α-klotho. The N-terminal region of FGF23 contains the FGF homology domain, while the C-terminal allows interaction with the cofactor α-klotho. Although FGF receptors (FGFRs) are widely distributed, the physiological actions of FGF23 are quite limited, due to the restricted pattern of expression of α-klotho, a protein with homology to β-glycosidase, and which exists as both a secreted or membrane-bound protein. Thus, FGF23signaling is most apparent in tissues that coexpress FGFR1c plus α-klotho, notably the proximal and distal convoluted tubule, and to a lesser extent the parathyroids, the pituitary, cardiac sinoatrial cells, placenta, skeletal muscle, urinary bladder, aorta, pancreas, testis, ovary, and colon.
Excess FGF23 production results in hypophosphatemia, reduced serum levels of 1,25(OH)2D and elevated PTH levels, and impaired bone and cartilage mineralization. FGF23 deficiency, on the other hand, results in hyperphosphatemia, elevated 1,25(OH)2D levels, suppressed PTH, and soft tissue calcification. Calcitriol increases FGF23 while FGF23 inhibits 1-α-hydroxylase activity. In mouse models, both overexpression and exogenous administration of FGF23 decreased renal expression of Npt2a and Npt2c, decreased renal phosphate reabsorption, decreased calcitriol production, and lowered serum phosphate. Current models suggest that FGF23 induces phosphorylation of ERK1/2 and serum/glucocorticoid kinase 1 (SGK1) in proximal tubule cells, and that SGK1 in turn phosphorylates NHERF-1, leading to internalization and degradation of NaPi-2a and NaPi-2c. PTH signals through activation of PKA and PKC, which also leads to phosphorylation of NHERF-1. Absence of α-Klotho causes an FGF23 deficient state with hyperphosphatemia, increased 1,25(OH)2D, and tissue calcification.26 Excess production of FGF23 is common in chronic renal failure, and recent studies suggest that elevated circulating levels of FGF23 may be the principal cause of myocardial damage and other adverse outcomes in these patients, due to effects that are unrelated to the physiologic actions of FGF23 on bone and mineral metabolism.
FGF23 is an important modulator of phosphate homeostasis in normal individuals, as there is a positive relationship between changes in dietary phosphate and circulating levels of FGF23.27 Other factors that appear important in tumor-induced osteomalacia include secreted frizzled related protein 4 (sFRP-4) and MEPE,28 but their roles in normal physiology have received limited attention.
Calcitonin is a 32-amino acid peptide synthesized and secreted by the parafollicular, or C cells, of the thyroid gland. The gene for calcitonin is composed of six exons and five introns, and the gene transcript can undergo alternative tissue-specific RNA processing to produce two different peptides (Figure 7-12). In the thyroid, the major product of the calcitonin gene is calcitonin, whereas in the brain, it is the calcitonin gene-related peptide (CGRP).
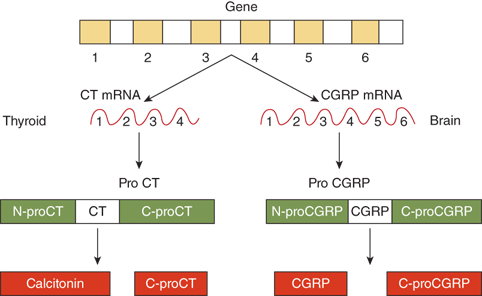
In all species, calcitonin is composed of 32 amino acids with a 1-7 amino terminal disulfide bridge and a carboxy-terminal prolinamide residue. The whole peptide is necessary for biological activity. Nonmammalian forms of calcitonin have greater potency than mammalian forms, even when tested in mammalians. Calcitonin is synthesized as a large precursor with an N-propeptide flanking the amino terminal end and a C-propeptide flanking the carboxy terminus (see Figure 7-12). Proteolytic processing of procalcitonin cleaves these flanking peptides. C-proCT is cosecreted with calcitonin in equimolar amounts, and radioimmunological measurement of the peptide may have clinical value in monitoring patients with medullary thyroid carcinoma.
Human calcitonin has a plasma half-life of 5 to 10 minutes. The kidney represents the predominant site of metabolism and subsequent clearance of the circulating hormone. Thus, the plasma calcitonin concentration maybe elevated in patients with renal failure.
The secretion of calcitonin is directly regulated by the plasma concentration of calcium. When blood calcium rises acutely, plasma calcitonin increases proportionally. Conversely, an acute decrease in plasma calcium produces a corresponding decrease in plasma calcitonin. However, the effects of chronic hypercalcemia and hypocalcemia are more complex; for example, normal, elevated, and decreased levels of calcitonin have been reported in hyperparathyroidism. Chronic hypocalcemia may result in increased storage of calcitonin in C cells, but does not appear to have any consistent effect on calcitonin secretion. Calcitonin is also released in response to the gastrointestinal factors gastrin, pentagastrin, cholecystokinin, and secretin and to neuroendocrine factors such as the α-agonist isoproterenol and the α-antagonist phentolamine. However, the physiological significance of these factors in regulation of calcitonin secretion remains unclear. Notably, most studies in humans fail to demonstrate an increase in plasma calcitonin after calcium-rich meals unless plasma calcium increases.
Similar to many other peptide hormones, calcitonin mediates its effects via activation of adenylyl cyclase with consequent increases in intracellular cAMP. The major biological effect of calcitonin is to decrease bone resorption by inhibiting osteoclast activity. In addition, calcitonin may reduce osteoclast formation and maturation. However, athyroid patients and patients with tumors that secrete calcitonin (eg, medullary thyroid carcinoma) lack clinical abnormalities in either bone histomorphometry or calcium homeostasis. In long-term in vitro experiments, the diminished inhibition of calcium resorption from skeletal tissue likely arises from desensitization of the calcitonin receptor.
Calcitonin circulates in several forms, including monomeric, dimeric, and larger forms of the hormone. The plasma concentration of monomeric calcitonin is very low in healthy children and adults (~10 pg/mL). Thus far, the clinical use of calcitonin measurements in plasma has been largely limited to detection of increased secretion in patients with medullary thyroid carcinoma.
Neonatal hypocalcemia is the most typical form of hypocalcemia encountered by the pediatrician and is particularly common in the neonatal intensive care unit. A fall of total calcium (adjusted for serum albumin concentration) below 7.5 mg/dL (1.75 mmol/L) or of ionized calcium below 2.5 mg/dL (0.63 mmol/L) is considered hypocalcemia in newborns. By contrasts, in infants 3 months of age and younger, hypocalcemia is defined as total serum calcium less than 8.8 mg/dL or ionized calcium less than 4.9 mg/dL (1.22-1.4 mmol/L). The relationship of the total calcium concentration to the ionized calcium concentration is atypical in preterm infants, and measurement of the ionized calcium concentration, when performed rapidly on a fresh blood sample, will provide the most accurate assessment of serum calcium concentration in these young patients.
In order to meet the high demand for minerals required by the developing skeleton, the fetus maintains higher blood calcium and phosphorus levels than present in the maternal circulation. This high demand is accomplished through active transport of calcium across the placenta by a calcium pump in the basal membrane that maintains a 1:1.4 maternal to fetal calcium gradient. A midregion fragment of PTHrP produced by the parathyroid glands and many other tissues is largely responsible for regulating the calcium gradient, but intact parathyroids and PTH are also required for maintenance of normal fetal levels of calcium (as well as magnesium and phosphate), and lack of parathyroid glands and PTH causes a greater fall in fetal serum calcium concentration than lack of PTHrP. The role of calcitriol in fetal physiology is not completely understood, but it appears to play a role in skeletal development.
Fetal circulating calcium levels, as reflected in the cord blood total serum calcium concentration, increase with advancing gestational age, and the “term” fetus is hypercalcemic relative to maternal concentrations of serum calcium. With birth the infant is abruptly disconnected from the placenta and the maternal calcium supply and, thus, becomes entirely dependent on intestinal absorption of dietary calcium and skeletal calcium reserves to maintain serum calcium levels. During this transition, the response of the parathyroid gland to falling levels of ionized calcium is somewhat deficient, resulting in a physiologic nadir in neonatal serum calcium levels within the first 2 days of life. The ability of the parathyroid glands to respond to the relative hypocalcemic challenge of birth depends on gestational age. Term infants typically achieve normal levels of serum calcium during the second week of life. Circulating concentrations of ionized calcium tend to be higher in neonates (range 1.0-1.5 mmol/L) than in older children or adults (range 1.12-1.32 mmol/L). In some newborns, secretion of PTH and/or responsiveness to PTH is not adequate to maintain normal serum levels of calcium (and phosphorus), suggesting delayed maturation of the parathyroid glands or proximal renal tubules, respectively.
The newborn has an especially high degree of adaptability for absorption of calcium depending on the amount of calcium present in the diet. Mature human milk contains 34 mg of calcium per 100 mL, and standard commercial formulas contain approximately 50 mg/100 mL. While absorption of calcium from human breast milk is greater than that from infant formulas, calcium absorption is enhanced regardless of diet when the infant receives adequate amounts of vitamin D. While neither human nor cow’s milk contains appreciable amounts of vitamin D, commercial infant formulas are fortified with vitamin D (400 IU per quart). Nevertheless, fewer than 50% of infants who are fed commercial formula will receive 400 IU of vitamin D per day. Hence, the American Academy of Pediatrics recommends that all infants under 12 months of age, regardless of whether they are fed human milk or commercial infant formula, receive a daily vitamin D supplement of 400 IU as soon as feasible. Preterm infants, and infants born to mothers with poor vitamin status, are at increased risk of vitamin D deficiency, and may have congenital rickets or hypocalcemia.
Inadequate supply or absorption of calcium can also cause hypocalcemia. The term infant is able to absorb and retain more calcium than preterm infants. Nonetheless, in preterm infants, as calcium intake increases, so does the intestinal absorption of calcium. This is particularly relevant, as the calcium content of human milk and standard infant formulas will not provide the preterm infant with sufficient calcium to meet the needs of the still developing skeleton, which has a calcium accretion rate of 100 to 150 mg/kg/day during the third trimester. Hence, inadequate dietary calcium is a significant contributor to low bone mineralization in preterm infants. Fortunately, intestinal maturation and responsiveness to vitamin D is accelerated by preterm delivery, and calcium absorption is not a limiting issue for the preterm infant. While lactose increases calcium absorption, possibly by reduction of the intestinal pH, the high phosphate content of infant formula can reduce calcium absorption through formation of nonabsorbable calcium phosphate salts.
In infants, as in older children, hypocalcemia causes neuromuscular irritability, which in the newborn may manifest as myoclonic jerks, “twitching,” exaggerated startle responses, and seizures. Apnea, cyanosis, tachypnea, tachycardia, vomiting, laryngospasm, or heart failure may also be seen. Markedly reduced ionized calcium concentrations may be associated with prolongation of the Qo-Tc interval on the electrocardiogram and decreased cardiac contractility. However, many infants with low calcium levels will lack specific symptoms or signs, and hypocalcemia may only be identified by blood chemistry studies. Thus, the pediatrician must be alert to possible hypocalcemia when unusual signs or symptoms are present, particulary in the preterm infant or infant of a diabetic mother.
Early neonatal hypocalcemia occurs within the first 3 days of birth and may be regarded as an accentuation of the physiologic fall in the plasma calcium concentration that takes place in all newborn infants in the first 2 to 3 days of life (Figure 7-13). Early neonatal hypocalcemia apparently results from insufficient release of PTH by immature parathyroid glands or inadequate responsiveness of the renal tubule cells to PTH. An exaggerated rise in calcitonin secretion in premature infants may contribute. Prematurity, low birth weight, hypoglycemia, maternal diabetes mellitus, difficult delivery, and respiratory distress syndrome are common associated findings. Hypomagnesemia has especially been observed in newborn infants of diabetic mothers, but it is usually mild, transient, and unlikely to play a prominent role in the pathophysiology of neonatal hypocalcemia in these infants. A more severe form of transient neonatal hypoparathyroidism and tetany occurs in infants who were exposed to maternal hypercalcemia in utero, as intrauterine hypercalcemia suppresses parathyroid activity in the developing fetus and presumably leads to delayed responsiveness of parathyroid glands to hypocalcemia after birth.
Classically, infantile tetany occurs after the first week of life in both full-term and preterm infants who are fed commercial infant formula or a cow’s milk-based formula. The high phosphate content of cow’s milk and many infant formulas can reduce calcium absorption in the intestine. In addition, many infants are unable to exrete the high phosphate load and develop hyperphosphatemia, which in turn directly reduces serum calcium levels and inhibits renal production of 1,25(OH)2D, which further reduces intestinal absorption of calcium. Because human milk is relatively low in phosphate, breast-fed infants rarely, if ever, develop late hypocalcemia. Hyperphosphatemia, and consequent hypocalcemia, may also arise from phosphate enemas.
Congenital hypoparathyroidism, which may be transient or permanent, can also account for neonatal hypocalcemia in some infants. In other infants, parathyroid secretion of PTH appears to be appropriate but relative resistance of the immature kidney to PTH reduces renal excretion of phosphorus. These latter biochemical features strongly resemble those of pseudohypoparathyroidism (PHP), but in contrast to genetic forms of PHP that are associated with defects in the GNAS gene, infants with this transient form of PTH resistance show normal nephrogenous cyclic AMP responses to administered PTH. Rarely, late-onset hypocalcemia may occur as a result of infantile hypomagnesemia or vitamin D deficiency, or maternal hypercalcemia or hypermagnesemia. The serum 1,25(OH)2D concentration can be low, normal or, in infants with elevated PTH levels, slightly elevated.
In most infants, neonatal hypocalcemia is transient, and calcium homeostasis becomes normal within 1 to 4 weeks. If hypocalcemia and hyperphosphatemia persist beyond 4 weeks, other causes of parathyroid dysfunction such as congenital hypoparathyroidism or specific deficiency of vitamin D or magnesium deficiency should be suspected (see the section “Hypocalcemia in Children and Adolescents”).
Symptomatic hypocalcemia should be treated with intravenous calcium, preferably as 10% calcium gluconate, administered as 1 to 2 mL/kg over 10 minutes. If intravenous catheters are used, they should not be placed near the heart because of the danger of cardiac arrest from rapid infusion of calcium. Extravasation of 10% calcium gluconate is injurious to the soft tissue and causes severe necrosis with precipitation of calcium in the injured tissue. Because intravenous calcium rapidly equilibrates within the body, most infants will become hypocalcemic again within several hours of this initial calcium infusion. Hence, additional therapy will be needed, including in some cases protracted treatment with intravenous calcium. Intravenous calcium is best administered as a continuous infusion (1-3 mg/kg/h of elemental calcium), but when excess fluid volume is a concern, calcium may be administered by intravenous bolus infusion every 4 to 6 hours. In infants with accompanying hypomagnesemia, the plasma magnesium concentration usually increases spontaneously as the plasma concentration of calcium returns to normal.
To maintain normal plasma calcium concentrations, oral calcium lactate or gluconate in a dosage of 75 mg of elemental calcium/kg/day in neonates and 50 mg/kg/day in older infants should be added. Vitamin D supplementation (400 IU per day) should be given, with higher doses in infants with vitamin D deficiency. In some cases, particularly when hypocalcemia is severe or suspected to be permanent, additional therapy with oral calcitriol (starting with 25-50 ng/kg/day) may be necessary. Formula-fed infants with typical late-onset hypocalcemia and elevated serum levels of phosphorus will benefit greatly from supplementation of artificial formulas with sufficient calcium to achieve a high (3:1-4:1) calcium/phosphorus ratio. Calcium lactate powder is 13% calcium by weight whereas calcium gluconate is 9% calcium. This is most easily accomplished using an infant formula containing low amounts of phosphate or breast milk. One useful approach has been to supplement a low phosphorus formula such as Similac PM 60/40 (11.2 mg calcium and 5.5 mg phosphorus per oz). For example, 5 oz of Similac PM 60/40 contains 1.4 mmol (56 mg) of calcium and 0.90 mmol (28 mg) of phosphorus, which corresponds to a molar Ca:P ratio of 1.6:1. In order to achieve a desired 4:1 Ca:P ratio, one needs 2.2 mmol of calcium (88 mg), which could be achieved by addition of 220 mg of calcium carbonate to 5 oz of formula. As treatment decreases the plasma phosphate concentration, the plasma calcium returns to normal over several days.
Intravenous calcium can usually be discontinued within 24 hours as oral therapy becomes effective, but withdrawal of intravenous calcium should be guided by repeated plasma calcium determinations, and, ideally, done gradually by halving the intravenous dosage successively for 2 days before discontinuation. Once the infant has maintained normal serum levels of serum calcium and phosphorus on oral therapy, the calcium supplement is reduced in steps, not stopped abruptly, as the phosphate level may rise precipitously leading to a fall in the calcium concentration to tetanic levels (Box 7-1).
Box 7-1. When to Refer
Hypocalcemia
Appropriate referral to the endocrinologist or other specialist should be made for hypocalcemia that is not attributable to a significant intercurrent illness or intervention or that is persistent. Such disorders include:
Severe vitamin D deficiency
25-α-hydroxylase deficiency
1-α-hydroxylase deficiency
Vitamin D receptor defects
Hypoparathyroidism
Pseudohypoparathyroidism
Gain-of-function mutations in calcium-sensing receptor
The clinical presentation of hypocalcemia can vary from an asymptomatic biochemical finding to a life-threatening condition. The manifestations of hypocalcemia are due primarily to enhanced neuromuscular excitability (tetany) of the central and peripheral nervous system, and in general reflect the level or rate of decline of the circulating level of ionized rather than total calcium. However, in most cases determination of the total serum calcium level can provide a useful guide to the ionized calcium level, and when plasma protein levels are normal, symptoms and signs of hypocalcemia are common when total calcium concentrations are approximately 7 to 7.5 mg/dL (1.75-1.87 mmol/L). In addition, the signs and symptoms of hypocalcemic tetany can be amplified by other electrolyte abnormalities, particularly hypomagnesemia and hypokalemia. Patients with chronic hypocalcemia sometimes have few, if any, symptoms of neuromuscular irritability despite markedly depressed serum calcium concentrations. By contrast, patients with acute hypocalcemia frequently manifest many symptoms of tetany. Most patients with hypocalcemia will have some mild features of tetany, including circumoral numbness, paresthesias of the distal extremities, or muscle cramps. Symptoms of fatigue, hyperirritability, anxiety, and depression are also common.
Latent or manifest tetany is the hallmark of hypocalcemia in children. Muscular pain, stiffness, and cramps are early manifestations. A typical attack of tetany begins with tingling sensations in the fingertips, and sometimes in the feet. The muscles then feel tense and subsequently spasm. The hands are most commonly involved producing the classic carpal spasm, that is, adduction of the thumb and extension of the fingers. Spasm in the feet is less common with plantar flexion of the toes, arching of the feet, and contraction of the calf muscle. The muscle spasm causes pain, which may be severe. Tetany attacks may be provoked by prolonged muscular exercise, emotional stress, or febrile illness. Spasm, including laryngospasm, can also be precipitated by hyperventilation leading to alkalosis and reduction of ionic calcium concentration.
The Chvostek, Trousseau, and Erb signs are indicative of hyperirritability and may be useful in the clinical diagnosis of latent tetany. The Chvostek sign is elicited by tapping the facial nerve 1 to 2 cm anterior to the earlobe with a fingertip. The muscle twitch in response to the stimulus can be graded as follows: twitching of upper lip at the corner of mouth only (grade 1), progress of twitching to include alae nasi (grade 2), and to include contraction of orbicularis oculi (grade 3). A grade 1 response may occur in somewhat more than 25% of normal children and in 10% to 30% of adults with normal serum calcium levels. Trousseau sign is evoked by inflating a blood pressure cuff on the upper arm above systolic pressure for up to 3 minutes to produce carpal spasm. Trousseau sign is present if carpal spasm occurs after compression of the nerves in the upper arm. In the presence of hypocalcemia, the neuroischemia caused by application of pressure to the upper arm induces sufficient irritability to yield a positive response: flexion of the wrist and metacarpophalangeal joints, extension of the interphalangeal joints, and adduction of the digits. The Erb peroneal sign is the flexion and eversion of the foot when the peroneal nerve is tapped just behind the head of the fibula. All of these signs can be absent even in patients with definite hypocalcemia.
Seizures resembling epilepsy may occur, and hypocalcemia can worsen an underlying primary seizure disorder. There, the serum calcium concentration should be measured as part of the routine evaluation of patients with seizures. When hypocalcemia occurs in the setting of chronic hypoparathyroidism calcifications may occur within the basal ganglia and diffuse calcification can occur at the gray-white matter interface of the cerebrum. These lesions are evident on CT scans of the brain. Although these calcifications are irreversible they only rarely cause extrapyramidal movement disorders or impair cognitive function.
Papilledema and pseudotumor cerebri may be found in long-standing hypoparathyroidism or may develop soon after institution of treatment. The papilledema is moderate in degree and is not accompanied by hemorrhage or impaired vision. Following treatment the papilledema usually begins to subside within a few days of normocalcemia, but it may take several weeks to disappear. Lenticular cataracts are a common complication of chronic hypocalcemia. They first appear as discrete punctate or lamellar opacities in the cortex, separated by a clear zone from the capsule. These ocular manifestations are irreversible but normalization of the plasma calcium arrests further progression.
Hypocalcemia delays ventricular depolarization and prolongs the heart rate-corrected Q-T and ST intervals on the electrocardiogram. A 2:1 heart block and, rarely, congestive cardiac failure may develop. Dry and scaly skin and brittle and fissured nails are ectodermal complications that occur in certain forms of hypoparathyroidism (see below). Eruption of teeth may be delayed and the formation of enamel may be irregular and hypoplastic.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


