Differentiation therapy
Sai-Juan Chen, MD, PhD  Xiao-Jing Yan, MD
Xiao-Jing Yan, MD  Guang-Biao Zhou, MD
Guang-Biao Zhou, MD  Zhu Chen, PhD
Zhu Chen, PhD
Overview
Abnormal differentiation is one of the main features of human cancers, especially in hematological malignancies. Many aberrant genetic and epigenetic factors have been shown to disrupt the regulation of cell differentiation in a variety of cancers and play an important role in oncogenesis. Differentiation therapy refers to the application of therapeutic agents selectively targeting the key molecules involved in the process of cell differentiation, leading to the restoration of normal cellular homeostasis and the eventual clearance of cancer cells. Over the past four decades, investigations of the molecular mechanisms underlying cancer cell differentiation arrest or blockage have allowed the identification of an array of drug targets. On the other hand, many physiological or pharmacological agents have been tested using in vitro and in vivo systems as the inducers of differentiation and maturation of cancer cells. The translational research in this field has gradually turned the differentiation therapy from a concept to clinical practices. The most successful model of cancer differentiation therapy is the development of synergistic targeted treatment of acute promyelocytic leukemia (APL) with all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO). This chapter discusses the basic theories of differentiation therapy and the clinical achievements of this therapeutic approach.
Molecular mechanisms underlying differentiation blockage in cancer
It has long been recognized that in human cancers, there exist some relationship between the clinical aggressiveness and their differentiation status, with poorly differentiated tumors being usually more aggressive. To understand the biology of differentiation arrest or blockage in cancer, it is necessary to elucidate how the related functions are regulated in normal cells and how they become disrupted in cancer cells. These studies are of importance for identifying meaningful therapeutic targets.
It is well established that pluripotent embryonic stem cells are capable of self-renewal and differentiating into various cells with specific functions in an organism. These cells can then develop into distinct cell types, tissues, and organs, which are under accurate regulation in a finely organized manner. A large body of evidence has been obtained that the underlying mechanisms entail the sequential action of cell-/tissue-specific or time-specific transcription factors (TFs) that activate or repress the differentiation-associated genes under appropriate conditions.1–3 The transcriptional expression of genes is also orchestrated by complex mechanisms designated as epigenetic regulation. Notably, many pathways and networks that regulate cell differentiation during normal development are affected by genetic and epigenetic abnormalities during tumorigenesis. As a result, cancer cell population generally bears an aberrant expression of regulators for embryonic morphogenesis and maintains a subset with properties of stem cells. Meanwhile, cancer cells often show defects in programs that keep cells in differentiated state (Figure 1).
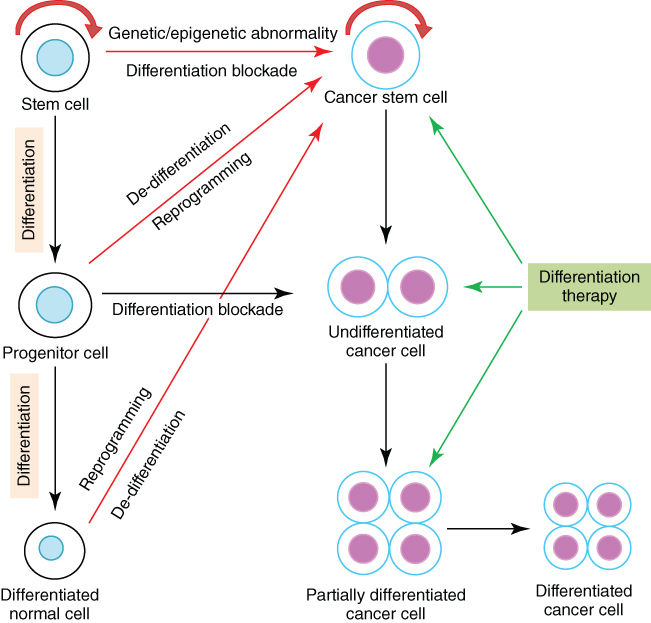
Figure 1 Differentiation blockade and differentiation therapy in cancer. In addition to self-renewal, normal stem cells are capable of differentiating into specific mature cells, while differentiation blockade may lead to the development of cancer. Cancer stem cells may originate from normal stem cells acquiring genetic and/or epigenetic abnormalities, or from normal progenitor or differentiated cells undergoing de-differentiation and reprogramming also due to genetic and/or epigenetic abnormalities. Differentiation therapy exerts therapeutic efficacy by inducing differentiation of cancer stem cells and undifferentiated cancer cells.
Transcriptional factors (TFs)
Cellular differentiation can be defined by a sequential process of acquisition of different functions, and cell fate is largely determined by control of gene expression through the combination of TFs and epigenetic modifiers.3, 4 TFs are nuclear proteins capable of DNA binding and trans-activating or trans-repressing the transcription of target genes. They are often assembled into multiprotein complex with cofactors (activators or repressors) and play decisive roles in regulating the gene expression profiles of stem/progenitor cells and determining their ability to differentiate into mature cell lineages.5 TFs are also essential for control of cell proliferation and programmed cell death (apoptosis). Recently, cellular reprogramming and induction of pluripotency across differentiated lineages have been achieved using combinations of TFs, which provide strong evidence for the key role that TFs play in the decisions of cell fate.6–11 In addition, that cell differentiation can be directed by lineage-specific TFs has also been supported by a large amount of experimental data.3, 5, 12 In contrast, abnormalities of differentiation-associated TFs due to dysregulated expression or aberrant functions imposed by gene point mutations or fusions may disturb the differentiation program of normal stem/progenitor cells.5 As a result, immature descendants with growth and survival advantages may be produced, eventually leading to the development of cancer.
The cancer-associated TFs can be divided into distinct classes according to structural and/or functional features,12 such as leucine zipper factors (bZIP, e.g., c-JUN), helix-loop-helix factors (bHLH, e.g., MYC), Cys4 zinc fingers [e.g., nuclear receptors (NRs), GATA factors], helix-turn-helix factors (HTH, e.g., HOX family, OCT-1/2), and Rel homology region (e.g., NF-κB). These TFs are considered as key regulators of cell proliferation, differentiation, and apoptosis, and their mutations and/or aberrant expression have been shown to play an essential role in malignant transformation. A special class of ligand-activated zinc finger proteins known as superfamily of NRs is worth particular attention, as they are major regulators in the initiation and/or maintenance of differentiated status of many cell/tissue types.13 This family contains the receptors for different ligands including steroid/thyroid hormones, retinoids, vitamin D3, and certain fatty acids. Notably, many of these receptors are subject to dysregulated expression or structural abnormalities in various cancers. These abnormalities subsequently lead to undesired activation of the normally hormone-regulated genes in a hormone-independent way, or even activation of some genes unrelated to hormone regulation, which will be discussed in detail in the section entitled “Agents targeting aberrant TFs.”
For long time, hematopoiesis has been at the frontline of research in the biology of normal and abnormal cell differentiation,5, 14, 15 owing to the discoveries of hematopoietic hormones and related signaling pathways and the specific TFs that regulate the fate of hematopoietic stem/progenitor cells (HSPC) (Figure 2). Each stage of myeloid and lymphoid differentiation shows particular TFs signatures that function in networks. For example, RUNX1, GATA1, C/EBPα, C/EBPβ, MYB, E2A, PAX5, TAL1/SCL, or PU.1 are turned on or turned off in an orchestrated manner to ensure blood cell lineage specification.3, 15 Some of these TFs were originally identified following the molecular characterization of gene point mutations or fusion in leukemia, such as C/EBPα in CEBPA mutation of acute myeloid leukemia (AML), RUNX1 in the RUNX1(AML1)–ETO fusion gene of t(8;21) AML, and SCL(TAL1) in t(1;14) T-cell acute lymphoblastic leukemia (ALL).16–18 Disruption of these hematopoietic TFs in leukemia blocks hematopoietic cells in their early stages of differentiation.
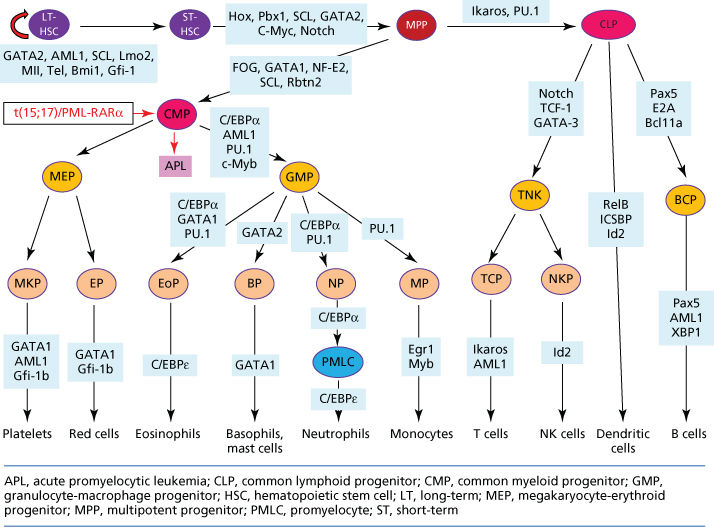
Figure 2 Signaling pathways and transcriptional factors (TFs) regulating the differentiation of hematopoietic stem/progenitor cells (HSPC). Differentiation block may result in leukemia, exemplified by development of acute promyelocytic leukemia (APL), a subtype of acute myeloid leukemia (AML) characterized by accumulation of abnormal promyelocytes in bone marrow/peripheral blood, the presence of t(15;17)/PML-RARα fusion gene, and severe bleeding syndrome. In fact, almost all the listed signaling molecules and TFs are subject to mutations (gene fusions, point mutations, gene amplifications/deletions) and/or dysregulation of expression in hematological malignancies.
The fact that abnormal TFs disturbing cell differentiation are “drivers” in oncogenesis has aroused interest for these proteins to be potential targets for therapeutic intervention. However, most current anticancer research activities are concentrated on cell-surface receptors because they offer a relatively straightforward way for drugs to affect cellular behavior, whereas agents acting at the level of transcription need to penetrate the cell membrane and enter into the nucleus. Efforts should be supported to design drugs affecting cancer-associated transcriptional patterns for re-establishing differentiation. Small molecule drugs directed against well-defined interacting sites between TFs and DNA or between TFs and other proteins in transcription machinery can be developed, so as to modulate the key TF target genes. On the other hand, the re-introduction of differentiation-related wild-type TFs with gene therapy might also serve as a strategy for cancer differentiation therapy.
Epigenetic modifiers
Epigenetic regulation constitutes crucial mechanisms governing the variability in gene expression, which is heritable through mitosis and potentially also meiosis, without changes of genomic sequence.19, 20 All cells of an organism share the same genome information but exhibit different phenotypes with diverse functions. This phenotypic diversity is due to distinct gene expression patterns among different cell types. It is well established that chromatin states are primary determinants for the turn-on or turn-off of genes. The assembly and compaction of chromatin require multiple regulatory mechanisms, including DNA methylation (cytosine methylation and hydroxymethylation) or demethylation, post-translational modifications of histones (phosphorylation, acetylation, methylation, and ubiquitylation), and noncoding RNA-mediated pathways.20–22 A large number of enzymes have been identified to be regulatory factors in the mechanisms, such as DNA methyltransferases (DNMTs) and DNA demethylases (DNDMs), histone deacetylases (HDACs) and histone acetyltransferases (HATs), and histone methyltransferases (HMTs) and histone demethylases (HDMs) (Figure 3a and b). The different modifications of DNA or histones are closely interrelated and can both reinforce each other and inhibit each other. Many histone-modifying enzymes are components of cofactor complexes and function cooperatively with TFs to modulate gene expression (Figure 3c).
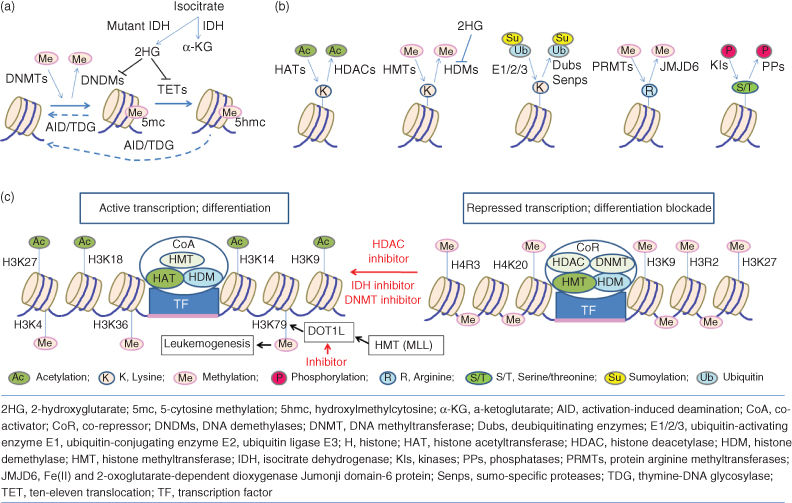
Figure 3 Epigenetic regulation involved in the cell differentiation and tumorigenesis. (a) DNA methylation (cytosine methylation and hydroxymethylation) are regulated by different enzymes DNMTs, TETs, and IDH. (b) Post-translational modifications of histones include phosphorylation, acetylation, methylation, and ubiquitylation or sumoylation, which are regulated by a variety of factors HATs, HDACs, HMTs, HDMs, and so on. Notably, most of these epigenetic modifier genes have been found to be mutated in human cancers including solid tumors and hematological malignancies. (c) Gene expression is controlled in the promoter regions by a combination of chromatin modification and transcriptional factor (TF). In tumor cells, gene expression is silenced by condensing chromatin, methylating DNA, and deacetylating histones, which results the differentiation blockade. Inhibition of HDACs, DNMTs, and IDH may result in active transcription and differentiation of cancer cells. Some HMTs (e.g., MLL and related fusion proteins) may enhance H3K79 methylation by promoting DOT1L to activate transcription of some leukemogenic genes, and inhibitor of DOT1L may suppress the leukemogenic genes to treat leukemia.
Recently, new technological platforms have allowed comprehensive epigenomic map to be made in pluripotent and differentiated cells, which provide further support to the concept that cell differentiation is accompanied by dynamic features of chromatin states.22–25 Genetic studies in different model organisms have shown the importance of chromatin regulators in key developmental transitions and cell fate decision. Cancer cells are characterized by typical genome-wide aberrations at the epigenetic level, such as the hypomethylation, the hypermethylation of specific promoter regions, the histone deacetylation, the downregulation of microRNAs (miRNAs), and the dysregulation of certain components of epigenetic machinery.20, 21, 26 Recently, somatic alterations of many epigenetic modifiers have been identified as common genetic events in cancers including solid tumors and hematopoietic malignancies.27–30 In acute leukemia and myelodysplastic syndrome (MDS), besides the already known aberrations of mixed lineage leukemia (MLL1) located on 11q23, mutations in a subset of epigenetic modifiers, including other SET domain-containing proteins (MLL2, SETD2),31 tet methylcytosine dioxygenase 2 (TET2),32–34 isocitrate dehydrogenase (IDH1/IDH2),35 additional sex combs-like 1 (ASXL1),36, 37 enhancer of zeste homolog 2 (EZH2),38, 39 and DNA methyltransferase 3A (DNMT3A),40, 41 have been discovered. These mutations have been shown not only to contribute to the leukemogenesis but also serve as the biomarker of clinical prognosis since they often predict a poor overall survival (OS) of patients.28–30 The aberrations of epigenetic modification together with those of genetic regulation result in permanent changes in the gene expression patterns that confer a selective advantage to differentiation block, apoptotic deficiency, and uncontrolled cell growth. Because the epigenetic modifications are usually reversible, therapies targeting epigenetic modifiers hold the promise of being clinically effective.
Cancer stem cells (CSCs)
Most cancers are heterogeneous in terms of cell proliferation and differentiation status. In recent years, evidence has been accumulated to suggest the existence of less-differentiated, stem cell–like cells within the cancer cell population in solid tumors (lung, colon, prostate, ovarian, and brain cancers and melanoma) and leukemia.42–45 These cells are now referred to as cancer stem cells (CSCs) or tumor-initiating cells capable of self-renewal and giving rise to the entire cancer bulk.46 Indeed, a single leukemia-initiating cell (LIC) from human being can cause a full-blown AML in animal model.47 As a result, malignant cells in a given cancer are highly hierarchical, with the unique self-renewing CSCs at the top of the hierarchy, while all other cells are derived from differentiated CSCs (Figure 1).
Investigation of the properties of CSCs and their roles in disease mechanisms occupies a central position in cancer research nowadays. Generally speaking, CSCs represent the driving force behind cancer initiation, progression, metastasis, recurrence, and drug resistance.43, 48 It is nevertheless not easy to define the phenotypes of CSCs in that they are variable from one cancer to another and are affected by the initial transformation as well as different stages of neoplastic development. Currently, fluorescence-activated cell sorting using cell surface markers or intracellular molecules represents the common method to identify CSCs.42, 43 There are still problems to be tackled: CSC biomarkers are lacking in some cancers; not all CSCs express the markers or marker sets; some differentiated cancer cells also express the same marker sets as CSCs do; some currently used marker sets are not specific to CSC since they are also expressed by stem/progenitor cells in normal tissues. On the other hand, gene expression patterns may be of value because dysregulation of certain pathways or networks can be seen in CSCs but not in differentiated cancer cell compartments, which can eventually be used as new CSC markers. So far, PI3K/Akt, PTEN, JAK/STAT, TGF-β, Wnt/β-catenin, hedgehog, Notch, NF-κB, and Bcl-2 have been included into the pathways that are involved in the control of self-renewal and differentiation of CSCs.42, 43
Traditional cytotoxic chemotherapy (CT) or radiation therapy is designed to target cancer cells with rapid proliferative or dividing abilities. These therapies are usually unable to eliminate CSCs, which are in relatively quiescent state and divide slowly, and can consequently regenerate cancers and drive disease recurrence.48–50 It is believed that only when CSCs are eradicated can cancer patients be actually cured. In recent years, great attention has been given to the characteristics of CSCs by both scientific and medical communities because these cells provide a unique target for cancer treatment. CSCs-targeted strategy focuses on the elimination of CSCs by acting on specific surface molecules, signaling pathways, or microenvironments, which are indispensable for the maintenance of stem cells, or by inducing CSCs into the more differentiated state.4, 45, 48, 49 A number of existing agents have been investigated in this regard and high-throughput screening of chemical libraries has also been used to find novel CSC-targeted chemicals.51, 52
Potential cancer cell differentiation-inducing agents
Cancer differentiation therapy aims at using appropriate therapeutic agents to induce cell cycle arrest and a commitment to the cell differentiation program toward terminal cell division, specific function gaining, and ultimately apoptosis.53 This concept applies relatively clearly to hematologic malignancies, whereas “authentic” differentiation therapy for solid tumors is sometimes difficult to define and often refers to the processes that convert malignant cells to a more benign phenotype, accompanied by diminished proliferative and metastatic abilities and the expression of mature cell markers.4 Interestingly, some initially defined differentiation inducers have turned out to be compounds targeting oncoproteins, while some newly developed targeting therapeutics have been found to trigger significant differentiation of cancer cells. Thus far, about 80 agents of differentiation induction have been identified and biosynthesized, and most of them are under investigations in vitro or on animal models, among which only a fraction have been used in clinical practice.
Agents targeting aberrant TFs
As discussed previously, TFs play a powerful role in physiological cellular differentiation and can be abnormally regulated or functionally disturbed in cancers. The modulation and/or restoration of the functions of differentiation-associated TFs constitute one of the main strategies for cancer differentiation therapy. Here, we introduce some differentiation-induction agents, which specifically target hormone-regulated TFs.
Retinoids
Retinoids are a group of vitamin A derivatives. Retinoic acids (RAs) are the physiologically existing active metabolite of retinol, which regulate a wide range of biological processes including development, differentiation, proliferation, and apoptosis in normal or cancer cells.13, 54–56 The most important isomers of RAs are all-trans-retinoic acid (ATRA), 13-cis-retinoic acid (13-cRA), and 9-cis-retinoic acid (9-cRA), which have different spectrums of biological activity and ligand affinity.57, 58 Synthetic retinoids include bexarotene and fenretinide. The effects of RAs on cell differentiation are exerted mostly via their NRs. Two types of receptors, the retinoic acid receptors (RARs, with three major isoforms RAR-α/-β/-γ) and the retinoid X receptors (RXRs, with three main isoforms RXR-α/-β/-γ), mediate RA signaling as heterodimer RAR/RXR.13, 56 These receptor heterodimers are able to bind, at genomic level, to the specific motifs defined as retinoic acid response elements (RAREs) on the promoters of target genes. RAR/RXR differentially activates or represses gene transcription while recruiting co-activators (CoA) or co-repressors (CoR) complexes. These different modes of action depend on the presence or absence of the ligands, RAs. Physiological concentrations of RA (1 pmol/L) are able to release CoR (such as N-CoR/SMRT complex and the chromatin remodeling complexes, e.g., HDACs, DNMTs) from the RAR/RXR and recruit CoA (such as p300/CBP complex with HAT activity), leading to the transcriptional activation of genes involved in cell differentiation (e.g., C/EBPα and PU.1), growth, and apoptosis.56, 59–65
Retinoid signaling is often disrupted during carcinogenesis, suggesting that restoration of this pathway may be a viable option for cancer treatment.54, 55, 58, 66 The effects of RA-induced differentiation are particularly observed on promyelocytic leukemia cell-like cell lines such as HL60 and NB4, or fresh primary APL (acute promyelocytic leukemia) cells.67, 68 These cells can undergo terminal differentiation to morphologically and functionally mature granulocytes after incubation with pharmacological concentrations (0.1–1 μmol/L) of RAs, especially ATRA.67 Myeloblasts from other AML subtypes are generally not affected by RAs, whereas only limited differentiating effects of retinoids are seen on cell lines such as THP-1 (monoblastic leukemia), K562 (blastic phase of CML), HEL (erythroleukemia), and U937 (monocytic leukemia).59, 68 Until now, the most successful clinical practice of differentiation therapy is the use of ATRA in APL, which will be discussed in details in the following section.
Retinoids have been reported to induce the differentiation of many solid tumor cell lines in vitro and suppress carcinogenesis in tumorigenic animal models, including human teratoma, melanoma, neuroblastoma, osteosarcoma, and rhabdomyosarcoma.57, 69, 70 In thyroid carcinoma cell lines, retinoids were shown to induce the expression of type I iodothyronine-5′-deiodinase and sodium/iodide symporter, which are the thyroid differentiation markers.71–73 In early clinical studies, patients with poorly differentiated, inoperable thyroid carcinoma with lacking or insufficient iodine uptake were treated with 13-cRA at oral doses of 0.3–1.5 mg/kg/day over 5 weeks and up to 9 months. The results showed an increased radio iodide uptake in 20–40% of patients.73–75 In some of these trials, afluorine-18 fluorodeoxyglucose positron emission tomography (18 F-FDG PET) was used for the evaluation of therapy with 13-cRA in thyroid cancer. These data indicate retinoids as promising differentiation therapy agents for the treatment of thyroid cancer.
In addition to induction of differentiation, retinoids exert antitumor effects through other modes of action: repressing cell proliferation, promoting cell apoptosis, and inhibiting angiogenesis and metastasis.56, 57 Notably, retinoids are used effectively in the treatment of certain cutaneous premalignancies and malignancies with good results.76 Bexarotene, the first synthetic highly selective RXR agonist, has been shown to induce apoptosis of cutaneous T-cell lymphoma (CTCL) cells with downregulation of its receptors and of survivin, an inhibitor of apoptosis.77
In clinical practice, systemic retinoids are approved by the US Food and Drug Administration (FDA) for the treatment of APL and CTCL.
Vitamin D compounds
Vitamin D is known to have a variety of actions at cell proliferation and differentiation.78 These actions are mainly mediated by vitamin D receptor (VDR), a member of NR superfamily. While performing physiological functions, VDR forms heterodimer with RXR. The VDR/RXR heterodimer binds to vitamin D response elements (VDREs) in the promoter regions of target genes.79 In the absence of physiologically active form of vitamin D, 1,25 dihydroxy vitamin D3 [1,25(OH)2D3](1,25D), the VDR/RXR heterodimer recruits co-repressors including HDACs and results in transcriptional repression.66 However, once the ligand binds to VDR/RXR, the resultant conformational change of the receptor heterodimer triggers the transcriptional activation of the target genes. It has been demonstrated that the recruitment of several coactivators to VDR/RXR is indispensable for this trans-activation. The VDR also directly interacts with a number of other proteins (e.g., β-catenin) and regulate their activities.80 Besides, 1,25D can trigger “rapid responses” via activating membrane initiated signaling pathways, such as MAPK pathways, the lipid signaling pathways, or PI3K/AKT pathway.79, 81, 82
Similar to the differentiation-inducing effects of retinoids, 1,25D has been shown to dispose differentiating actions on various types of human AML cells,78 including HL-60, U937, NB4, THP-1, and KG-1. Animal model studies have shown that 1,25D strongly promotes cell differentiation and thereby significantly prolongs the survival of mice transplanted with leukemic cells. However, the results of clinical trials are unsatisfactory. Neither 1,25D nor vitamin D analogs (VDAs) show clear beneficial effects on patients with AML or MDS.83, 84 An important issue for these failures could be the heterogeneous nature of the diseases. The clinical utility of 1,25D has also been limited by the severe toxicity of its therapeutic doses, primarily due to fatal drug-induced hypercalcemia.85
Some types of solid tumors, such as colon, breast, and prostate cancers and osteosarcoma, were also reportedly to respond to in vitro applied VDAs.86, 87 Nevertheless, little is known about how VDAs mediate their antiproliferation or pro-differentiation activity.86 Moreover, the results of clinical trials of VDAs across a variety of cancers have been disappointing with regard to efficacy.88 Thus, the routine recommendation of vitamin D for cancer treatment is premature.
PPARγ agonists
Peroxisome proliferator-activated receptor gamma (PPARγ) is also a member of the NR superfamily.89 PPARγ forms heterodimer with RXR, which then binds to PPARγ response elements in the promoter regions of target genes. It is well established that PPARγ is important in the regulation of proliferation, differentiation, and apoptosis in many cell types.89 Synthetic PPARγ agonists have been developed, including drugs used in the treatment of type 2 diabetes [e.g., troglitazone, rosiglitazone (TGZ)] and nonsteroidal anti-inflammatory agents (e.g., indomethacin).59, 69
Activation of PPARγ with agonists can suppress the growth and induce the differentiation of several types of tumors in vitro and in animal models for colon, breast, prostate, and thyroid cancers, liposarcoma (LPS), pituitary adenomas, and acute leukemia.90–92 Antitumor effects of PPARγ agonists may also be associated with regulation of cell cycle, apoptosis, and expression of tumor suppressor genes (TSGs, e.g., PTEN and BRCA1).59, 93, 94 Besides, there has been evidence for “off-target” effects of PPARγ agonists, which is independent on the PPARγ receptor activity.59
Clinical trials of PPARγ agonists have been carried out in several human malignancies. A few studies have reported that TGZ could induce the differentiation of cancer cells in patients with LPS.95 It was initially found that terminal adipocytic differentiation was induced in a few patients with intermediate to high-grade LPS. However, such responses and clinical benefit could not be confirmed in a subsequent trial.96 Clinical investigations in breast, prostate, colon, lung, and thyroid cancers and LPS did not reveal a meaningful benefit of TGZ therapy.59, 73, 96–98 Although the clinical effects of PPARγ agonists have not been convincing, their low toxicity profile and potential additive or synergistic effects combined with other anticancer agents such as retinoids make them reasonable candidates in combination therapy trials.
Agents targeting epigenetic modifiers
It has been shown that the aberration of epigenetic regulation and TFs governing differentiation occurs in many cancers. Recurrent somatic alterations in epigenetic modifiers are also observed in solid tumors and hematological malignancies. Hence, there has been substantial interest in preclinical and clinical studies for epigenetic regulating agents to be considered as differentiation inducers.99 A large amount of epigenetic agents are currently under investigation and a few have received FDA approval. Promising results have been already yielded in certain cancer types.
DNMTs inhibitors
DNA methylation is the most characterized epigenetic modification and is described as a stable epigenetic marker.100 The enzymes catalyzing DNA methylation are known as DNMTs. As hypermethylation of TSGs and over-expression of DNMTs have been established as the key factors in carcinogenesis, demethylating agents seem to be especially promising as anticancer drugs.101, 102 To date, 5-azacytidine (5-Aza) and 5-aza-2′-deoxycytidine (decitabine) are the most successful DNMTs inhibitors, already approved by FDA for the treatment of patients with MDS or AML, and 5-Aza has also been approved by the FDA and the European Medicines Agency (EMA) for the treatment of chronic myelomonocytic leukemia (CMML).101 The mode of action seems dose-dependent and a “dual mechanism” has been identified: the cytotoxicity and/or the inhibition of cell proliferation at high doses, and the DNA hypomethylation effect on the induction of gene expression at low doses.101, 102 Numerous studies have described terminal differentiation of AML cells treated with 5-Aza or decitabine.102–104 Moreover, it has been demonstrated that decitabine-induced killing of myeloid leukemia cells is independent on DNA damage and apoptosis.102 In some solid tumor cells, 5-Aza or decitabine induces cancer cell apoptosis and senescence through multiple mechanisms.105
Recent clinical studies indicated that repeated exposure at low doses (decitabine: 20 mg/m2 for 5 days every 28 days; 5-Aza: 75 mg/m2 for 7 days every 28 days) to these agents can induce DNA demethylation with a better antineoplastic effect than when used at higher doses (decitabine: 45 mg/m2 for 3 days every 42 days).102, 106–109 In MDS patients, 5-Aza induces an overall response rate of 20–30% and significantly improves survival compared with standard care, while decitabine induces cytogenetic remission (Cyto-CR) rates of 35–50% at optimal doses and prolongs survival when compared with historical controls.102, 106–108, 110, 111 Clinical trials of DNMTs inhibitors in combination with other agents have also obtained promising results in solid tumors such as nonsmall-cell lung cancer and refractory ovarian cancer.105, 112, 113 Therefore, rational use of the existing DNMTs inhibitors are to be encouraged and efforts to develop new compounds targeting more selectively DNMTs and with less toxicities should be continued.
HDAC inhibitors
As enzymes catalyzing the deacetylation of histones, HDACs are involved in the control and regulation of cell growth and differentiation. Abnormal expression or mutations of HDACs, together with aberrant histone acetylation patterns, have been found in a broad range of cancer types.114 HDACs have thus become a novel class of targets in cancer therapy (Figure 3c). Histone deacetylase inhibitors (HDACIs) comprise diverse compounds such as suberoylanilide hydroxamic acid (SAHA, vorinostat) or valproic acid (VPA). It has been reported that HDACIs promote growth arrest, differentiation, and apoptosis of many solid tumor cells (e.g., lung, prostate, thyroid, and breast cancers, chondrosarcoma, and glioblastoma) and cell lines of hematopoietic malignancy, and exhibit only minimal effects on normal tissues.69, 115–121 For instances, SAHA and VPA are able to induce in vitro myeloid differentiation and early apoptosis of Kasumi-1 AML cells, which express AML1-ETO. Similar effects were also reported in a murine xenograft tumor model.
Inspired by preclinical data, many researchers have investigated the effects of HDACIs in clinical trials. Early study of SAHA showed efficacy in a panel of hematologic diseases.122 The compound has been successfully applied to the treatment for refractory CTCL and becomes the first HDACI approved by FDA for this disease. A pilot study combining VPA and ATRA in eight refractory or high-risk AML patients demonstrated clinical benefits in seven cases, with myelomonocytic differentiation of circulating blasts. Although promising results have been obtained in hematological malignancies, clinical data for HDACIs in patients with solid tumors are inadequate.123 In future, the effects of the combination of HDACI with conventional cancer treatment should be explored in solid tumor.
DOT1L inhibitor
MLL gene is one of the epigenetic modifier genes identified through molecular cloning of chromosomal translations involving 11q23. This gene encodes a SET domain histone methyltransferase, which catalyzes the methylation of lysine 4 of histone H3 (H3K4) at specific gene loci.124 MLL is essential for fetal and adult hematopoiesis in that it regulates the expression of HOX genes and some cofactors key to the expansion and differentiation of HSPCs.125–127 Abnormalities of MLL gene account for over 70% of infant leukemia and approximately 10% of adult AML, and are correlated to a particularly worse clinical outcome.128 In addition to MLL translations, MLL partial tandem duplication (PTD) is also reported as a recurrent genetic defect. In the chimerical genes, the catalytic SET domain of MLL is lost and the fusion partners are capable of interacting with a histone H3K79 methyltransferase DOT1L.129, 130 As a result, fusion products not only retain gene-specific recognition elements within the MLL moiety, but also gain the ability to recruit DOT1L to these locations. The ectopic H3K79 methylation due to recruitment of DOT1L leads to enhanced expression of leukemogenic genes, such as HOXA9 and MEIS1.131 Of particular interest is that DOT1L contributes to the transforming activity of MLL fusion and thus functions as a catalytic driver of leukemogenesis.
It has been proposed that inhibition of DOT1L may provide a pharmacologic basis for therapeutic intervention in MLL-related leukemia (Figure 3c). In vitro and in vivo experiments have demonstrated that EPZ-5676, a specific inhibitor of DOT1L, can selectively kill cells carrying on the MLL gene translocation by causing cell cycle arrest and promoting cell differentiation.132 Data of phase 1 clinical trial of EPZ-5676 for the treatment of refractory or relapsed MLL rearranged acute leukemia patient reported that the compound was generally safe and well tolerated (NCT01684150). Eight of the 34 MLL-related patients showed biological or clinical responses,133 including two complete responses (CRs) and one partial response (PR). Other DOT1L inhibitors such as EPZ4777 and SYC-522 also have been documented to promote differentiation and apoptosis.134–136 Interestingly, bioinformatics analysis suggested that DOT1L may play a role in the pathogenesis of breast cancer. In vitro study has shown that DOT1L inhibition selectively inhibited proliferation, self-renewal, and metastatic potential of breast cancer cells and induced cell differentiation.137
Inhibitors of isocitrate dehydrogenase (IDH)
Somatic mutations of the metabolic enzyme IDH have recently been reported in human glioma, melanoma, thyroid cancer, chondrosarcoma, and AML.35, 138–140 All mutations are located at arginine residues in the catalytic pockets of IDH1 (R132) or IDH2 (R140 and R172) and thus confer on the enzymes a new activity: catalysis of α-ketoglutarate (α-KG) to 2-hydroxyglutarate (2-HG).140, 141 High concentrations of 2-HG, known as an “oncometabolite,” have been shown to inhibit α-KG-dependent dioxygenases, including histone and DNA demethylases, which are main modifiers of the epigenetic state of cells (Figure 3a).140–143 It has been suggested that cancer-associated IDH mutations may induce a block of cellular differentiation to promote tumorigenesis and therefore may be potential targets of differentiation therapy.143, 144 Indeed, it has been shown that a selective IDH1/R132H inhibitor (AGI-5198) induces the expression of genes associated with gliogenic differentiation and triggers astroglial differentiation of IDH1/R132H mutant (mIDH1) cells.145 Moreover, blockade of mIDH1 suppresses the growth of glioma cells with IDH1/R132H, but not those with wild-type IDH1 in vitro or in IDH1/R132H glioma xenograft mice. It is worth noting that AGI-6780, a small molecule that potently and selectively inhibits the tumor-associated mutant IDH2/R140Q, has also been developed. AGI-6780 is capable of inducing differentiation of TF-1 erythroleukemia and primary human AML cells in vitro.146 These findings provide proof-of-concept that inhibitors targeting mutant IDH1 or IDH2 may have potential applications as cancer differentiation inducers.
Recently, promising results of early phase clinical studies on AG-120 (the first inhibitor of mutated IDH1) and AG-221 (the inhibitor of the mutated IDH2 protein) were reported (https://www.clinicaltrials.gov/).144 AG-120 was well tolerated and showed encouraging clinical efficacy in patients with advanced IDH1-mutation positive AML. The compound seemed able to reduce 2-HG levels in diseased cells to normal levels and allow leukemia cells to mature into granulocytes. Among 14 evaluable cases out of 17 patients, 7 patients responded to the drug, with 4 CR (Daniel Pollyea, the 26th European Organization for Research and Treatment of Cancer Symposium). Consistent with the clinical observation, AG-120 reduces intracellular 2-HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo (56th ASH meeting, abstract 70455). The ongoing phase 1 study of AG-221 as a single agent in patients with IDH2-mutant positive advanced hematologic malignancies also showed a favorable safety profile as well as durable clinical benefits (56th ASH meeting, abstract 70721). Out of 48 patients enrolled, 20 patients achieved objective responses, including 12 CRs and 8 PRs. Responses were durable, with durations up to 8 months. Interestingly, AG-221 induces differentiation and impairs self-renewal of IDH2-mutant leukemia cells in murine models of IDH2-mutant leukemia and primary human AML xenograft models (56th ASH meeting, abstract 70656 and 76334).
Actually, the studies on cancer differentiation inducers gradually take momentum. Of particular interest is the model disease APL, where differentiation-inducing agents have been demonstrated to exert a curative effect in the great majority of patients. Besides, some other differentiation-induction agents have shown promising effects in preclinical experiments and clinical trials. Differentiation therapy for cancers, although still in its relative infancy, has given hope to millions of cancer patients.
APL as a successful model of cancer differentiation therapy
APL is the M3 subtype of AML, characterized not only by distinct clinical manifestations but also by unique molecular pathogenesis and effective target-based differentiation therapy.
Clinical treatment strategies in APL
APL was first described in 1957 as the most malignant form of acute leukemia.147 Patients with APL show specific features: a very rapid fatal natural course of only a few weeks’ duration, an accumulation of abnormal promyelocytes in bone marrow, and a severe bleeding syndrome due to disseminated intravascular coagulation (DIC) or hyperfibrinolysis. In the era before ATRA-based therapy, only 23–35% of the APL patients could have a 5-year survival by CT.148 Inspired by the Chinese philosophy of educational approach in health maintenance and the Western medical science of induction of cancer cell differentiation, scientists from Shanghai Institute of Hematology (SIH) carried out the research on leukemia differentiation therapy since the late 1970s.148
In 1980, a group from United States published the results of in vitro terminal differentiation of HL-60 cells upon the effect of 13-cRA.67 Clinical improvement or CR after treatment with 13-cRA in a few isolated cases of APL were then reported, accompanied by maturation of promyelocytes with disappearance of signs of coagulopathy.149–151 The SIH group, on the other hand, was focused on the potential therapeutic effects of ATRA. The first clinical trial of ATRA was reported in 24 APL cases (16 newly diagnosed and 8 anthracycline refractory cases) who were treated with ATRA alone.152 CR was achieved in all patients and the evidence of in vivo blast differentiation with Auer’s rods in polynucleated granulocytes was documented. The results have been quickly confirmed by a large amount of clinical practices around the world.59, 148 ATRA therapy became the standard for induction of newly diagnosed APL patients, which was then further improved stepwise through the effective combination regimen of ATRA with CT. Moreover, the roles of ATRA and CT in consolidation or maintenance therapy have been verified to reduce the incidence of relapse. By using this protocol, 5-year OS was up to about 70%.69, 148, 153–155 The routine prescription of ATRA is 45 mg/m2/day, while the recommendation dosage of ATRA in the SIH is 25 mg/m2/day. The reason to reduce the initial dose of ATRA is to avoid ATRA toxicity but to achieve the same therapeutic effects as the conventional dosage.156
Even though the ATRA/CT combination was considered very efficient anti-APL therapy, up to about 30% of APL patients still ended with relapse.157–159 The introduction of arsenic trioxide (ATO) since the early 1990s has further changed the clinical landscape of APL. In vitro studies showed that ATO exerts dose-dependent effects on APL cells including differentiation and apoptosis, although the differentiation is only a partial one compared with that induced by ATRA.160 Clinically, refractory or relapsed APL after ATRA/CT therapy as well as newly diagnosed APL could achieve CR, suggesting an absence of cross-resistance between ATRA and ATO.161 Therefore, new therapeutic strategies have focused on minimizing CT and administering ATRA plus ATO as primary therapy. Indeed, the ATRA-/ATO-based protocol yielded a much longer survival in newly diagnosed APL compared with therapy with ATRA or ATO alone.148, 162, 163 These outcomes were even not influenced by FLT3 mutation status, which was a poor prognostic factor in APL patients treated with ATRA and CT.164 Combination of ATRA and ATO have obtained about 95% of CR rate and 5-year DFS and OS were near 90%.162 Of note, the combination therapy without CT was associated with very high 2-year EFS and OS rates (97% and 99%, respectively) in nonhigh-risk APL by Sanz Score (Table 1).158, 165 Thus, in some newly updated authoritative guidelines, the ATRA-/ATO-based therapy has been the first-line strategy for APL patients. Although consensus has not yet been reached on the details of specific consolidation or maintenance therapy, it has been widely accepted that ATRA, ATO, and anthracycline-based CT should be given sequentially. The recommendation of APL treatment is listed in Tables 2 and 3.
Table 1 Sanz score for APL risk stratification
| Characteristic | Low risk | Intermediate risk | High risk |
| WBC (×109/L) | ≤10 | ≤10 | >10 |
| Platelets (×109/L) | >40 | ≤40 | ≤40 |
Source: Data from Ref. 165.
Table 2 The recommendation of APL treatment (in China)
| Nonhigh-risk APL | High-risk APL | |
| Induction therapy | ATRA 25 mg/m2/d × 28–42 or until clinical remission | ATRA 25 mg/m2/d × 28–42 or until clinical remission |
| ATO 0.16 mg/kg/d × 28–35 or until BM remission | ATO 0.16 mg/kg/d × 28–35 or until BM remission | |
| HU 20–40 mg/kg/d (when WBC > 10 × 109/L) | IDA 6–8 mg/m2/d × 3 or DNR 45 m/m2/d × 3 | |
| Consolidation therapy | DA: DNR 45 mg/m2/d × 3 + Ara-C 100–200 mg/m2/d × 7 | DA or (DNR + Ara-C 1.5–2.5 g/m2/d × 3) |
| MA: MTZ 6–8 mg/m2/d × 3 + Ara-C 100–200 mg/m2/d × 7 | MA or (MTZ + Ara-C 1.5–2.5 g/m2/d × 3) | |
| HA: HHT 2–3 mg/m2/d × 3 + Ara-C 100–200 mg/m2/d × 7 | HA or (HHT + Ara-C 1.5–2.5 g/m2/d × 3) | |
| Maintenance therapy | ATRA 25 mg/m2 × 28 | ATRA 25 mg/m2 × 28 |
| ATO 0.16 mg/kg × 28 | ATO 0.16 mg/kg × 28 | |
| 6-MP + MTX: 6-MP 100 mg/d on days 1–7, 15–21 MTX 20 mg/d on days 1, 8, 15, 21 | 6-MP + MTX: 6-MP 100 mg/d on days 1–7, 15–21 MTX 20 mg/d on days 1, 8, 15, 21 | |
| 5 cycles of sequential use of ATRA, ATO, and CT | 5 cycles of sequential use of ATRA, ATO, and CT |
Abbreviations: Ara-C, cytarabine; ATO, arsenic trioxide; ATRA, all-trans-retinoic acid; BM, bone marrow; HU, hydroxyurea; IDA, idarubicin; DNR, daunorubicin; MTZ, mitozantrone; HHT, homoharringtonine; 6-MP, 6-mercaptopurine; MTX, methotrexate.
Source: Data from Ref. 166.
Table 3 The recommendation of APL treatment (NCCN guideline 2015)
| Induction therapy | Consolidation therapy | |
| High-risk APL | ATRA 45 mg/m2/d until clinical remission + DNR 50 m/m2/d × 4 + Ara-C 200 mg/m2/d × 7 | ATO 0.15 mg/kg/d × 5 days/week × 5 weeks × 2 cycles, then (ATRA 45 mg/m2/d × 7 + DNR 50 mg/m2/d × 3) × 2 cycles |
| ATRA 45 mg/m2/d until clinical remission + DNR 60 m/m2/d × 3 + Ara-C 200 mg/m2/d × 7 | (DNR 60 mg/m2/d × 3 + Ara-C 200 mg/m2/d × 7), then (DNR 45 mg/m2/d × 3 + Ara-C 1.5–2 g/m2 Q12h × 3) | |
| ATRA 45 mg/m2/d until clinical remission + Idarubicin 12 mg/m2/d on days 2, 4, 6, 8 | (ATRA 45 mg/m2/d × 15 + IDA 5 mg/m2/d × 3 + Ara-C1g/m2/d × 4), then (ATRA 45 mg/m2/d × 15 + MTZ 10 mg/m2/d × 5), then (ATRA 45 mg/m2/d × 15 + IDA 12 mg/m2/d × 1 + Ara-C 150 mg/m2 Q8h × 4) | |
| ATRA 45 mg/m2/d × 1–36 + Idarubicin 6–12 mg/m2/d on days 2, 4, 6, 8 + ATO 0.15 mg/kg/d × 9–26 | (ATRA 45 mg/m2/d × 28 + ATO 0.15 mg/kg/d × 28), then (ATRA 45 mg/m2/d × 7 d/2 wk × 3 + ATO 0.15 mg/kg/d × 5 d/wk × 5 wk) | |
| Nonhigh-risk APL | ATRA 45 mg/m2/d until clinical remission + ATO 0.15 mg/kg/d until BM remission | ATO 0.15 mg/kg/d × 5 d/wk × 4 wk/8 wk × 4 cycles + ATRA 45 mg/m2/d × 2 wk/4 wk × 7 cycles |
| ATRA 45 mg/m2/d until clinical remission + DNR 50 m/m2/d × 4 + Ara-C 200 mg/m2/d × 7 | ATO 0.15 mg/kg/d × 5 d/wk × 5 wk × 2 cycles, then (ATRA 50 mg/m2/d × 7 + DNR 50 mg//m2/d × 3) × 2 cycles | |
| ATRA 45 mg/m2/d until clinical remission + DNR 60 m/m2/d × 3 + Ara-C 200 mg/m2/d × 7 | (DNR 60 mg/m2/d × 3 + Ara-C 200 mg/m2/d × 7) × 1 cycle, then (DNR 45 mg/m2/d × 3 + Ara-C 1 g/m2 Q12h × 4) × 1 cycle | |
| ATRA 45 mg/m2/d until clinical remission + Idarubicin 12 mg/m2/d on days 2, 4, 6, and 8 | (ATRA 45 mg/m2/d × 15 + IDA 5 mg/m2/d × 3), then (ATRA 45 mg/m2/d × 15 + MTZ 10 mg/m2/d × 5), then (ATRA 45 mg/m2/d × 15 + IDA 12 mg/m2/d × 1) |
Abbreviations: Ara-C, cytarabine; ATO, arsenic trioxide; ATRA, all-trans-retinoic acid; BM, bone marrow; DNR, daunorubicin; HHT, homoharringtonine; IDA, idarubicin; 6-MP, 6-mercaptopurine; MTZ, mitozantrone; MTX, methotrexate; NCCN, US National Comprehensive Cancer Network.
Source: Data from http://www.nccn.org/professionals/physician_gls/f_guidelines.asp.
The conventional administration of ATO is the intravenous route. An oral formulation of ATO was also found to be active in relapsed APL and has subsequently been evaluated in upfront management. Another formulation of arsenic in advanced clinical investigation is tetra-arsenic tetra-sulfide (AS4S4) and an oral As4S4-containing formula named the Realgar–Indigo naturalis formula (RIF) in traditional Chinese Medicine was used in APL patients.167, 168 No significant differences were noted between the RIF and intravenously administered ATO groups with regard to the CR rate, the 3 year of OS or the 2 years of DFS at 2 years.169, 170 These two trials have established the feasibility and safety of prolonged oral arsenic administration, but long-term safety data are still required. Oral arsenic formulations are not currently available in the United States or Europe.
As the prognosis of APL patients is getting much better, the issues of early mortality [related to bleeding, differentiation syndrome (DS), or infection] and relapse [including central nerve system (CNS) relapse] becomes increasingly important. In patients with clinical and pathologic features of APL, ATRA should be started upon first suspicion of APL without waiting for genetic confirmation of the diagnosis.166 Early initiation of ATRA may prevent the lethal complication of bleeding. DS is a common complication in the ATRA or ATO treatment, with a frequency being around 25% both in United States and in Europe,171, 172 but relatively rare in East Asian populations (5–10%).154, 162 Signs and symptoms of this syndrome include hyperleukocytosis, dyspnea with interstitial pulmonary infiltrates, peripheral edema, unexplained fever, weight gain, hypotension, and acute renal failure. The mortality could be down to 3% or lower if DS is recognized early and treated promptly with CT and high dose of dexamethasone (10 mg twice daily).172, 173 In addition, patients with a high white blood cell count (WBC > 30 × 109/L) at diagnosis may benefit from prophylactic steroids. A common practice to avoid CNS prophylaxis (methotrexate 5–10 mg, cytarabine 40–50 mg, and dexamethasone 5 mg) for APL patients are recommended, 4–6 times for nonhigh-risk APL and 6–8 times for the patients with high WBC counts.166 Another issue is side effects of ATO because the compound has long been considered as a very strong poison. In fact, the protocol incorporating ATO proves to be quite safe. The common adverse effects such as minor bone marrow myelosuppression, hepatotoxicity, gastrointestinal reactions, and neurotoxicity can be controlled and are generally reversible without need of the discontinuation of the drug.162, 166 In very rare situation, clinically significant arrhythmias due to prolongation of the QT interval on the ECG are observed, which can be avoided with appropriate precautions and withdrawal of the medicine.174 At the same time, arsenic concentrations in the urine of patients who had ceased maintenance treatment for 2 years were below the safety limits recommended by government agencies in several countries or regions, whereas arsenic levels in plasma, nails, and hair were only slightly higher than those found in healthy controls.162
Leukemogenesis and therapeutic mechanisms of APL
It has been well known that PML-RARα resulting from t(15;17)(q22;q21) translocation is the key driver of APL leukemogenesis, which exerts dominant negative effects on RARα- and PML-related pathways.175 The fusion protein represses the transcriptional expression of target genes essential for hematopoietic differentiation and yields an increasing proliferation and self-renewal of LICs.175–177 The PML-RARα fusion protein binding to RXR co-receptor functions as a constitutive transcriptional repressor at RAREs of target genes through recruitment of CoR, which leads to the characteristic differentiation block.148, 176, 178 In normal cells, PML proteins multimerize to form multiprotein subnuclear structures called PML-nuclear bodies (PML-NBs).179 PML-NBs have been shown to play an important role in DNA damage repair, apoptosis, growth, senescence, and angiogenesis, and more recently, in the maintenance of HSPCs. In APL cells, PML-NBs are disrupted owing to the formation of PML/PML-RARα heterocomplex, interfering with the normal biological functions of PML.180 This mechanism probably cooperates with the disruption of RAR/RXR pathway to enforce the APL-specific differentiation block and acquisition of self-renewal, thus transforming the committed HSPCs into immortal, fully transformed LICs (Figure 4).
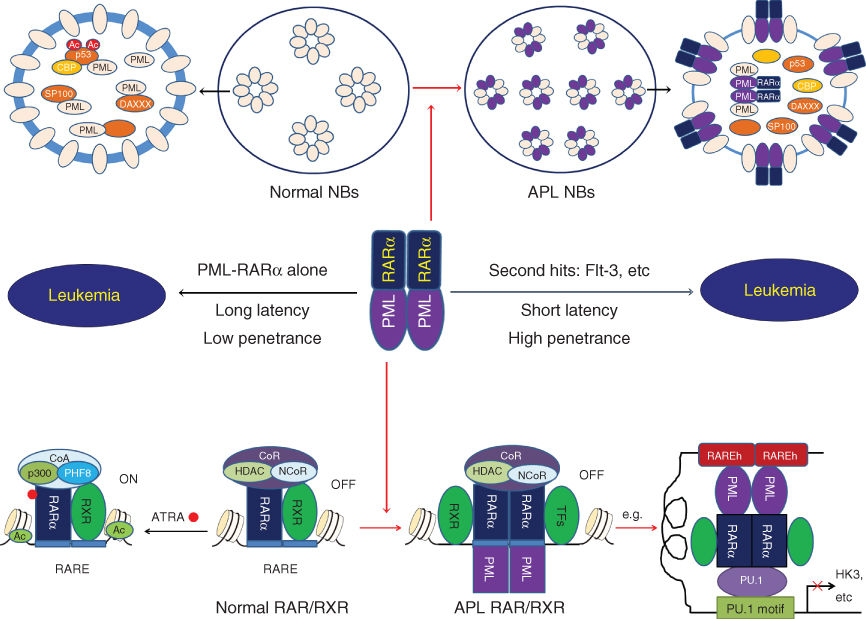
Figure 4 Molecular pathogenesis of APL. PML-RARα plays a key role in leukemogenesis of APL, which exerts dominant negative effects on RARα- and PML-related pathways. Under physiological condition, RAR/RXR heterodimers bind to the specific motifs defined as retinoic acid response elements (RAREs) on the promoters of target genes and differentially activates or represses gene transcription while recruiting co-activators (CoA) or co-repressors (CoR) complex depend on the presence or absence of the ligands, such as ATRA. The fusion protein binding to RXR co-receptor functions as a constitutive transcriptional repressor at RAREs of target genes through recruitment of CoR. In other hand, PML proteins multimerize to form multiprotein subnuclear structures called PML-nuclear bodies (PML-NBs) in normal cells. In APL cells, PML-NBs are disrupted owing to the formation of PML/PML-RARα heterocomplex.
Combining the discovery of PML-RARα in APL pathogenesis with special effective treatment of ATRA and ATO has pointed to a possible molecular mechanism underlying PML-RARα-specific therapy. Indeed, a large number of studies have revealed that ATRA and ATO act through distinct but complementary mechanisms, providing a biologic rationale for the two agents to be used in combination to achieve synergistic efficacy with low toxicity.
At pharmacological level (10−7–10−6 mol/L), ATRA binds to the ligand-binding domain (LBD) of RARα portion in the fusion protein and induces a conformational change of the chimerical receptor.181, 182 This leads to displacement of CoR complexes and recruitment of CoA complexes and clearance of PML-RARα from promoters of target genes, thereby restoring wild-type RARα function and subverting the differentiation block.63, 148, 178, 183 ATRA also leads to the degradation of PML-RARα oncoprotein,184–186 which may contribute to the response, and generate a restoration of nuclear architecture with reformation of PML-NBs. Re-establishment of RARα signaling allows differentiation of APL blasts and yields short-term disease management. Even though ATRA and anthracyclines are very efficient anti-APL therapies, APL patients can relapse, perhaps due to ATRA failing to affect LICs.
ATO exerts dose-dependent effects on APL cells in initial cellular and molecular mechanistic studies.160, 187 At low concentrations, ATO induces partial morphologic differentiation in APL cells, whereas at high concentrations, apoptosis induction occurs predominantly. Both effects are associated with a degradation of PML-RARα. ATO efficiently triggers the degradation of PML-RARα and PML through direct binding to the RBCC domain of PML moiety, which induces the conformational changes of PML proteins, leading to protein–protein aggregation and sumoylation of these proteins.188 In addition, ATO-induced oxidative stress promotes PML homodimerization by cross-linking PML via disulfide bonds.189 Sumoylated PML and PML-RARα recruit the ubiquitin ligase RNF4 and subject to proteasome-mediated proteolysis.190, 191 Besides, arsenic can act through other mechanisms independent of PML-RARα status, such as pro-apoptotic effects mediated by the mitochondria-mediated pathway, DNA damage, telomerase activity, autophagy, and so on.148, 192 Notably, ATO could induce LIC eradication in APL through several pathways, which may be a key factor in the success of ATO for APL patients.148, 177, 183, 192, 193 Sufficient and rapid degradation of PML and PML-RARα is required for LIC clearance and long-term disease eradication,194, 195 which can be obtained by ATO treatment. Arsenic can also facilitate elimination of LICs through inhibiting Notch pathway, antagonizing the Hedgehog–Gli pathway, and repressing NF-κB and β-catenin. The pleiotropic behaviors may be the reason why arsenic is very active in APL, as a single agent as well as in combination.
Because ATRA and ATO target respectively the C– and N-terminals of PML-RARα, enhanced degradation of PML-RARα oncoprotein might provide a plausible explanation for the superior efficacy of combination therapy in patients. Intriguingly, consistent with the mechanistic researches, genetic mutations in the LBD of the RARα moiety that interfere with ATRA binding or nuclear coregulators binding are observed in about 40% of relapsed APL patients treated with ATRA/CT,196–198 while genetic mutations in the PML moiety of the PML-RARα oncogene that probably impair the direct binding of ATO to PML-RARα in patients with clinical ATO resistance have been described after treatment with ATRA/ATO/CT.199, 200 A small number of APL patients harbor PLZF-RARα fusion gene resulting from t(11;17)(q23;q21), which are resistant to ATRA and ATO therapy.201, 202 These data further support that PML-RARα is the direct target of both ATRA and ATO. Several groups have shown that ATRA and ATO display a synergy in many pathways including TFs and cofactors, activation of calcium signaling, stimulation of the IFN pathway, activation of the proteasome system, cell cycle arrest, gain of apoptotic potential, downregulation of telomerase and telomere length, upregulation of cAMP/PKA activity, and enhanced arsenic uptake by APL cells through induced expression of cell membrane arsenic transporter (AQP9).148, 176, 203, 204 Recent studies have shown that the ATRA/ATO combination rapidly clears PML-RARα + LICs, resulting in APL eradication and dramatically prolong survival in murine models.177, 194 All these findings may contribute to the dramatically improved response from APL patients under the treatment of ATRA and ATO combination.
In conclusion, ATRA and ATO exert different but cross-linked actions on APL cells. ATRA mainly works through transcriptional modulation, and the main effects of ATO occur at the protein level, while both agents target PML-RARα. These data may further explain the lack of cross-resistance between the two agents (Figure 5). Therapy of APL with ATRA and ATO is to date the most successful example of cancer differentiation therapy, and key factor can be the synergistic targeting of the oncoprotein by the two agents. This scientific history may hence serve as a template for subsequent development of similar treatments in other leukemias and solid tumors. More importantly, the story of APL provides new way for thinking that differentiation therapy is essentially target therapy on the molecules affecting differentiation pathway and combination or synergistic targeting strategy is highly effective to eliminate CSCs.
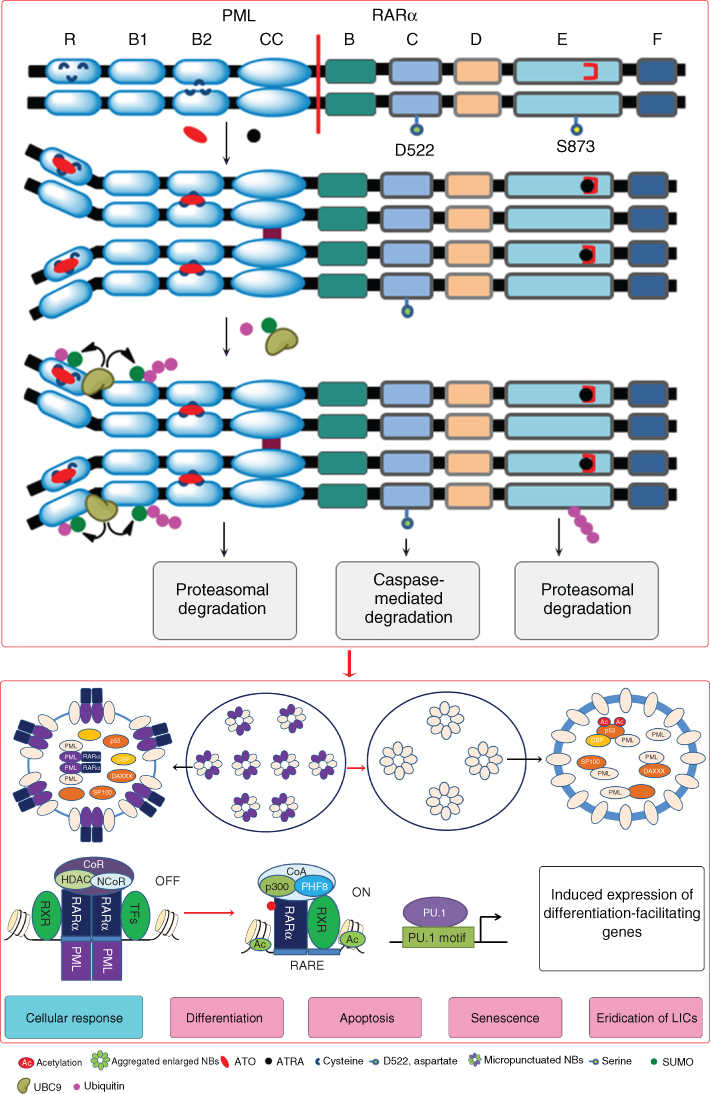
Figure 5 Therapeutic mechanism of APL. ATRA and ATO target respectively the C– and N-terminals of PML-RARα, enhancing degradation of PML-RARα oncoprotein. ATRA binds to the ligand-binding domain (LBD) of RARα portion in the fusion protein and ATO binds to the RBCC domain of PML moiety, which induces the conformational change and the consequential degradation of the fusion protein. The degradation of PML-RARα generates a restoration of nuclear architecture with reformation of PML-NBs. ATRA binding also leads to displacement of CoR complexes and recruitment of CoA complexes and clear PML-RARα from promoters of target genes, thereby restoring wild-type RARα function. The synergistic effects of ATRA and ATO result in a variety of cellular response including differentiation, apoptosis, and senescence of leukemic cells, and importantly, the eradication of leukemia-initiating cell (LIC).
Perspectives
Taken together, differentiation therapy against cancer has the potential to make tumor cells convert from a malignant path to a benign course, which has given hope to scientists and clinicians on a much better cancer treatment outcome. However, interest and application of differentiation-based therapy for solid tumors and hematological malignancies other than APL are just at the beginning. One major challenge is the complexity of histopathological subtypes and clinical stages of cancers, resulting in the absence of developmental models of cancer progression. The re-establishment of the genotype that characterizes the original tissue types and the morphological transformation of tumor cells to the normal cells may help to determine more successful differentiation induction. In addition, there have not yet been definite markers for evaluating the effects of differentiation inducers. Assessment of the precise therapeutic role of agents is often hampered by the difficulties in distinguishing in vivo cytotoxicity from differentiation. The classic evaluation of therapeutic responses mainly focuses on the shrinkage of the tumor mass, but this is not suitable for the response evaluation of differentiation therapy that just restores the differentiation program of tumor cells. Consequently, identifying accurate novel biomarker sets of response to differentiation therapy becomes urgent for clinical application. Recent evidence suggests that targeting of leukemia driver proteins other than PML-RARα can induce cell differentiation in some other types of AML and can eventually yield high-quality CR, paving the way of enlarging this approach in hematological malignancies. Nevertheless, solid tumors are still viewed as much more heterogeneous aberrant tissues than most leukemia, and much more complex molecular mechanisms are involved in their pathogenesis. Hence, the development of differentiation therapy in solid tumors may need more comprehensive approach to combine CT, immunotherapy, and differentiation agents in order to produce multiplied synergistic targeting effects.
Summary
Most human cancers show characteristics of abnormal cell differentiation, often coupled with dysregulation of proliferation and/or apoptosis. Cancer cells may be blocked at a particular stage of differentiation along with the involved cellular lineage, or they may differentiate into an inappropriate cell type. Hence, cancer differentiation therapy represents the approach aimed at the re-activation of endogenous differentiation programs or subverting differentiation/maturation blockage within cancer cells, often accompanied by the loss of malignant phenotype and the restoration, at least in part, of the normal phenotype.
The concept of differentiation therapy was pioneered by the work of Pierce and Verneyin 1961 when they observed differentiation of teratocarcinoma cells.205 In 1970s, important reports were made to demonstrate the differentiating capability of DMSO on erythropoiesis, the differentiation of neuroblastoma cells with some inducers, and the morphologic and functional maturation of leukemia cells induced by certain agents.59, 67, 206–208 Many substances have thus been shown to possess the potential of cancer differentiation inducer with in vitro cell line experiments. To trigger malignant cells to overcome their differentiation block and to enter the apoptotic pathways has become an elegant alternative to the therapies simply killing malignant cells. However, differentiation therapy had historically been hampered by many factors, especially due to the insufficient understanding of the pathways of normal cell differentiation and the much higher complexity of the process of cancer cell reversion to “normal” cells/tissues induced by differentiation therapy than the cytotoxic approaches.69
The advancement of differentiation therapy to a real successful clinical practice was not realized until the use of ATRA and ATO in the treatment of APL.53 This breakthrough has transformed APL from once a fatal disease to one of the most curable human cancers, with 5-year OS rates of 85–90%.148 In addition, over the past several years, progress has been made in understanding the differentiation pathways that are also cross-talking with those of the regulation of cell proliferation and survival, and the development of some targeted therapies on other types of leukemia has already yielded promising differentiation-inducing effects, which need to be further explored clinically.
This chapter focuses on the basic theories of differentiation therapy, and the clinical achievements of this novel therapeutic approach.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


