Differential Diagnosis of the Bleeding Patient
Jill M. Johnsen
Barbara A. Konkle
The hematologist is often faced with the question of whether a patient has an underlying bleeding disorder. This may be in the setting of unexplained bleeding, either spontaneous or with minor or major trauma or procedures. Or, the hematologist may be asked to evaluate a person’s risk of bleeding with an upcoming procedure, particularly in cases of a personal or family history of bleeding or screening laboratory results that suggest a bleeding risk. This chapter reviews key points to help determine whether an underlying bleeding disorder is present, first focusing on the history and physical examination. Basic screening laboratory tests usually available at the time of evaluation are reviewed, including the platelet count, prothrombin time (PT), also expressed as the international normalized ratio (INR), and activated partial thromboplastin time (aPTT). Laboratory tests useful in pinpointing specific hemostatic abnormalities are then discussed. Many of the disorders highlighted here are presented in more detail in other chapters in this text.
Bleeding disorders are classically divided into those of primary or secondary hemostasis. Disorders of primary hemostasis include those that affect initial platelet plug formation and typically result in mucosal bleeding, usually the result of a platelet or von Willebrand factor (VWF) disorder. Disorders of secondary hemostasis involve disorders of fibrin formation and often manifest as soft tissue or joint bleeding. [This is commonly due to specific coagulation factor deficiencies, notably factor VIII (FVIII) and factor IX (FIX) (hemophilia A and B, respectively)]. However, there is considerable overlap in symptomatology.1 Thus, although the pattern of bleeding is an important clue to its etiology, a comprehensive approach is necessary to correctly identify the bleeding patient’s underlying hemostatic defect.
THE BLEEDING HISTORY
From a detailed personal and family history, the clinician should have a good idea of whether (a) a bleeding disorder is present; (b) the disorder is inherited or acquired; (c) the pattern of bleeding suggests a primary or secondary hemostatic defect (or combination); and (d) a person’s propensity to bleed has been enhanced by an underlying medical condition or exposure to medications or dietary supplements.
A history of bleeding is the most important predictor of future bleeding risk. In evaluating a patient for a bleeding disorder, a history of hemostatic challenges should be assessed. Does the patient have a history of spontaneous or trauma- or surgery-induced bleeding? What is the nature and timing of the bleeding? Was the bleeding severe enough to necessitate medical intervention or treatment for acute or chronic blood loss?
A negative bleeding history is reassuring, but should not be used alone as the basis for dismissing a bleeding disorder, particularly if the person has never experienced a significant hemostatic challenge. Assessing a positive bleeding history can also be challenging, particularly in milder cases, as normal individuals can respond positively when questioned about specific signs or symptoms, particularly easy bruising or menorrhagia. Bleeding score questionnaires have been developed by several groups in an attempt to quantify bleeding in inherited bleeding disorders.2 These instruments may be useful as a guide to the type of questions to ask in a targeted bleeding history, but their utility in initial bleeding evaluations or scoring the severity of disease remains to be seen. Thus, a careful and thoughtful investigation of the bleeding history is critical in guiding any clinical evaluation of the bleeding patient.
Mucosal Bleeding
Epistaxis
Epistaxis is a common symptom, particularly in children and in dry climates, and may not reflect an underlying bleeding disorder. Clues that epistaxis is a symptom of an underlying bleeding disorder include lack of seasonal variation and bleeding of prolonged duration (>10 minutes), bleeding which requires medical attention, or frequent bleeding episodes.
Oral Mucosal Bleeding
Excessive bleeding or swelling after episodes of minor oral trauma may be a clue to an underlying bleeding disorder. Bleeding with eruption of primary teeth is seen in children with more severe bleeding disorders, such as moderate and severe hemophilia, but is uncommon in children with mild bleeding disorders. Spontaneous gum or cheek bleeding resulting in blood blisters, frankly bloody secretions, or profuse bleeding after tooth brushing in the absence of gum disease are also suggestive of an underlying bleeding disorder, especially disorders of primary hemostasis.
Dental extraction, particularly of molars or premolars, represents a significant hemostatic challenge. The limitations on wound closure imposed by the bony tooth socket and the presence of profibrinolytic enzymes in saliva and at the mucosal surface contribute to difficulties in achieving hemostasis. Excessive bleeding and/or recurrent bleeding after tooth extractions or other oral surgeries, or increased bleeding after dental cleaning, can be suggestive of an underlying bleeding disorder.
Gynecologic and Obstetrical Bleeding
Gynecologic and obstetrical bleeding are common presentations in women with underlying bleeding disorders (reviewed in Ref.33). Menorrhagia is a common symptom, but the history can be difficult as a complaint of heavy menses is subjective and
has a poor correlation with excessive blood loss. The diagnosis of menorrhagia is supported by anemia, soaking through a pad or tampon within 1 hour, or flooding at night.4 Additionally, women with underlying bleeding disorders are more likely to have other bleeding symptoms and are much more likely to have menorrhagia beginning at menarche than are women with menorrhagia due to other causes (reviewed in Ref.55).
has a poor correlation with excessive blood loss. The diagnosis of menorrhagia is supported by anemia, soaking through a pad or tampon within 1 hour, or flooding at night.4 Additionally, women with underlying bleeding disorders are more likely to have other bleeding symptoms and are much more likely to have menorrhagia beginning at menarche than are women with menorrhagia due to other causes (reviewed in Ref.55).
Postpartum hemorrhage is a common symptom in women with underlying bleeding disorders. This occurs most commonly in the first 48 hours after delivery but may also be manifest by prolonged, recurrent, or excessive bleeding after discharge from the hospital. Women with a history of postpartum hemorrhage are at an increased risk of recurrence in subsequent pregnancies. Women presenting with rupture of ovarian cysts along with intra-abdominal hemorrhage are also more likely to have an underlying bleeding disorder.3
Other Mucosal Bleeding
Gastrointestinal (GI) bleeding is usually due to underlying anatomic pathology and not a hemostasis defect. However, von Willebrand disease (VWD), particularly acquired VWD and types 2 and 3, can be associated with angiodysplasia of the bowel resulting in GI bleeding.6 True hematuria is more likely to be due to an underlying urologic or renal problem and not a bleeding diathesis.7 Rarely, recurrent hematuria of unclear etiology can occur in moderate and severe hemophilia.
Easy Bruising
The development of bruises (ecchymoses) after trauma is normal; however, an exaggerated response to trauma may be an indication of an underlying bleeding disorder. Ecchymoses presenting without known trauma, particularly on the trunk, and/or especially large (>3 cm in diameter) or palpable ecchymoses may be a sign of an underlying bleeding disorder. A lifelong history of significant ecchymoses suggests a congenital hemostatic defect, while the sudden development of new or multiple ecchymosis may reflect an acquired disorder. In pediatric and other vulnerable populations, ecchymoses can raise concerns for physical abuse, and the hematologist may be asked for an assessment of an underlying bleeding disorder (reviewed in Ref.88).
Minor Trauma
Bleeding after minor trauma is also normal. However a persistent history of excessive or prolonged bleeding after minor wounds, such as small sharp cuts from razors, paper, or knives, particularly bleeding which lasts >5 minutes or cannot be managed by the patient themselves, may suggest an underlying bleeding disorder. The hospitalized patient undergoes minor trauma with high frequency due to phlebotomy and the placement of intravascular catheters in addition to other procedures. The unexpected requirement for application of prolonged pressure or intervention at phlebotomy sites, vascular access sites, or after other minor procedures may be a sign of defective hemostasis.
Joint and Muscle Bleeds
Hemarthroses and spontaneous muscle or soft tissue hematomas are characteristic of moderate or severe congenital factor deficiencies, most commonly FVIII or FIX deficiency. It can also be seen in moderate and severe deficiencies of fibrinogen, prothrombin, and FV, FVII, and FX.1 Spontaneous hemarthroses occur rarely in other bleeding disorders except for severe VWD, which can be associated with FVIII levels <5%. Bleeding into a joint results in severe pain and swelling, as well as loss of function, but is rarely associated with discoloration from bruising around the joint. The first bleeding episode into a joint is accompanied by considerable swelling, but as bleeding recurs and joint destruction progresses, swelling may be less prominent while pain and loss of movement may worsen. In patients without a diagnosis of hemophilia, joint bleeding may go unrecognized as such by the patient or family. The clinical history, therefore, should specifically include questions about symptoms of severe joint pain and swelling.
Excessive Traumatic or Surgical Bleeding
Hemorrhage in the setting of severe trauma can be due to a combination of the trauma injury itself, surgical bleeding, medications, underlying comorbidities, and acquired coagulopathies, which can have components of both transfusion-related coagulopathy (discussed below) and trauma-related coagulopathy. Trauma-related coagulopathy itself is multifactorial and can result from shock and resulting tissue hypoperfusion and organ dysfunction, hypothermia, acidemia, dilution, consumption of clotting factors, and injured tissue-mediated activation of anticoagulant and fibrinolytic factors. Not surprisingly, trauma patients with underlying coagulopathies have worse outcomes.9
Significant surgical bleeding can occur during surgery or in the postoperative period. A comprehensive assessment of sites of bleeding, procedures, and local measures to stop the bleeding is critical. Different procedures pose different hemostatic challenges that must be considered in the bleeding evaluation. Tonsillectomy is a significant hemostatic challenge from both the surgery and mucosal injury. Bleeding may occur early after surgery or more than a week postoperatively with loss of the eschar at the operative site.10,11 Surgeries also inherently incur different degrees of blood loss in the normal patient. Some vascular or orthopedic procedures, such as spinal surgery, are bloody surgeries and can require transfusion support even in the hemostatically normal patient. On the other hand, other general surgeries, such as laparoscopic intra-abdominal procedures, can generally be performed with minimal blood loss. A number of major surgeries, such as coronary artery bypass grafting or aortic procedures, are often performed while the patient is exposed to one or more anticoagulant medications, which increases blood loss from the surgical sites and exacerbates bleeding from any other underlying anatomic defects.
Hemorrhagic events often occur in patients with comorbidities and/or multiple confounding factors. For example, the medical patient with liver failure (acquired factor deficiencies) being treated for infected ascites (risk for disseminated intravascular coagulation [DIC], antibiotic effects) may bleed briskly from esophageal varices (anatomic site) resulting in hypotension (tissue shock response) and a need for multiple transfusions (dilutional coagulopathy). However, even in cases instigated by a seemingly simple anatomic event, such as cleaning a penetrating wound with excessive bleeding requiring transfusion, factors can rapidly compound to a multifactorial coagulopathy. The approach to complex bleeding patient requires an open mind to reevaluate the history of the patient and periodic reassessments both for clinical and laboratory response to interventions and for new developments that may contribute to bleeding. Ongoing laboratory assessments in such patients should include at least a PT, aPTT, platelet count, and fibrinogen. Further laboratory
analyses will depend on laboratory results and underlying disorders. As patients with liver disease can have laboratory findings consistent with low-grade DIC, a diagnosis of DIC may require serial testing to show evidence of ongoing fibrinogen consumption.
analyses will depend on laboratory results and underlying disorders. As patients with liver disease can have laboratory findings consistent with low-grade DIC, a diagnosis of DIC may require serial testing to show evidence of ongoing fibrinogen consumption.
Patients with underlying bleeding disorders are at higher risk to bleed in the setting of trauma or surgery. The hematologist should take into account the nature of the trauma or procedures performed and consider the opinion of the surgeon in assessing if the patient is bleeding excessively for the hemostatic challenge.
Transfusion-Related Coagulopathy
Transfusion-related coagulopathy occurs in the setting of largevolume transfusion replacing acute blood loss (reviewed in Ref.109). The primary cause is dilution and failure to adequately replace plasma coagulation factors or platelets in proportion with the number of red cell units transfused. Replacement can be guided by closely following the platelet count, PT/INR (for plasma), or fibrinogen levels (for fibrinogen concentrates or cryoprecipitate). In high-volume, rapid transfusion, the need for these blood components can generally be anticipated by the number of red cell units infused. Massive transfusion also results in infusion of significant doses of citrate with the blood products. Iatrogenic hypocalcemia, reflected by low ionized calcium levels, may also contribute to clinical bleeding. Citrate chelates calcium, an essential cofactor for clotting and for multiple other essential biologic processes, including nerve conduction and muscle contractility.
Underlying Systemic Disease and Bleeding
Acquired bleeding disorders are commonly secondary to, or associated with, systemic disease. The clinical evaluation of a patient with a bleeding tendency must therefore include a thorough assessment for evidence of underlying medical disorders. Bruising or mucosal bleeding may be the presenting complaint in liver disease, severe renal impairment, hypothyroidism, and conditions causing bone marrow failure. Less prevalent disorders, such as paraproteinemias or amyloidosis, occasionally also present in this fashion. In DIC, presentation may be with symptoms of bleeding and/or microvascular thrombosis, and the underlying trigger for the consumptive coagulopathy is not always immediately obvious,12 such as in cases of occult malignancy.
Other Conditions Presenting with Signs or Symptoms of Bleeding
Medical conditions in which there is no coagulopathy or defect of primary hemostasis can present with signs or symptoms, which may suggest a bleeding disorder, but are instead caused by abnormalities of blood vessels or their supporting tissues. For example, in inherited collagen disorders, such as Ehlers-Danlos or Marfan syndrome, there is frequently a history of easy bruising or prolonged posttraumatic bleeding. However, other clues in the history and physical point to the underlying congenital defect, such as skin hyperelasticity or joint hyperextensibility and body habitus. Endocrine disorders (such as Cushing syndrome) or chronic exogenous corticosteroid exposure can result in changes in the skin and subcutaneous tissues that result in more pronounced subcutaneous bleeding in response to minor trauma. The bleeding is generally more superficial than in true ecchymoses. Senile purpura, common with advancing age, is also very superficial, usually occurs on sun-damaged skin, most commonly on the hands and forearms, and can also be mistaken for ecchymoses. Some autoimmune disorders can present with vasculitis reminiscent of petechiae and skin findings suggestive of bruising. Joint pain and swelling due to an acute arthritic flare, crystal arthropathy, or septic joint can mimic hemarthroses. These are often able to be differentiated from a true joint bleed by the history, an important distinction as suspicion for these other inflammatory conditions may lead to arthrocentesis, a procedure that generally should be avoided in patients with a hemarthroses related to an underlying bleeding disorder.
Medications and Dietary Supplements
A complete medication list, including both short-term and chronic medications and all herbal or supplement use, is essential and should be temporally correlated to the onset of bleeding. A common but sometimes overlooked contributor to bleeding is the use of aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs inhibit both cylooxygenase-1 (COX-1) and COX-2 pathways. Inhibition of COX-1 impairs primary hemostasis and can exacerbate bleeding from another cause or even unmask previously occult mild bleeding disorders. NSAIDs selective for the COX-2 enzyme have little platelet inhibitory effect and should not enhance or precipitate bleeding, but have instead been associated paradoxically with increased risk for thrombotic events.13 However, all NSAIDs can precipitate GI bleeding by direct action at the GI mucosa, which may then be more severe in patients with underlying bleeding disorders. The aspirin effect on platelet function is permanent and can persist for up to 7 days as assessed by platelet aggregometry, although it frequently returns to normal by 3 days after the last dose.14 The duration of the effect of other NSAIDs on platelets is shorter, because the inhibitor effect is reversible and depends on the half-life of the agent.
Other causes of drug-related bleeding range from the obvious, such as hemorrhage complicating thrombolytic therapy or bleeding in the setting of thrombocytopenia after chemotherapy, to the more diagnostically challenging, such as bleeding secondary to impaired platelet function in subjects on high-dose intravenous penicillin or caused by a combination of a qualitative platelet defect and moderate thrombocytopenia secondary to anticonvulsant therapy with sodium valproate (see Chapter XX for further discussion of drug-induced qualitative platelet defects). Furthermore, exposure to multiple medications contributes to the risk of adverse drug-drug or drug-supplement interactions, which can lead to altered drug levels and toxicity.
Use of alternative medicines and herbal supplements is commonly practiced and must be asked about specifically. Popular herbal supplements, such as gingko, ginseng, and garlic, are associated with altered hemostasis15 (Table 50.1). Fish oil or concentrated omega 3 fatty acid supplements can also impair platelet function. Herbal supplements may also alter hemostasis indirectly by interacting with other drugs, particularly warfarin. In addition to these concerns, loose regulatory oversight of the supplement industry raises concerns for quality control of the products consumed. Herbal supplements may contain variable (or no) detectable amounts of the active agents claimed on the packaging, while other unlisted ingredients can be present due
to poor manufacturing processes, misidentification, or substitution. NSAIDs are a common contaminant, particularly for those formulations claiming pain relief,15 and warfarin was repeatedly identified in formulations of PC-SPES, a dietary supplement.16
to poor manufacturing processes, misidentification, or substitution. NSAIDs are a common contaminant, particularly for those formulations claiming pain relief,15 and warfarin was repeatedly identified in formulations of PC-SPES, a dietary supplement.16
Table 50.1 Herbal supplements associated with increased bleeding | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Social History
The bleeding history must take into account the patient’s exposure to hemostatic challenges in their daily lives. This includes assessing both common daily activities and the history of the individual’s participation in sports or other vigorous physical activities. The lifestyle of an office worker or low-impact exerciser provides fewer opportunities for incidents of trauma compared to individuals who have physically demanding occupations or engage in contact sports. In the same vein, an infant who cannot walk would not be expected to exhibit the minor ecchymoses commonly found on the normal toddler. This history will help guide the clinician in assessing if bleeding symptoms, particularly ecchymoses or bleeding with minor trauma, seem excessive for the patient’s circumstances.
FAMILY HISTORY
Young age at presentation, prolonged duration of symptoms, failure of previous hemostatic challenges, and positive family history can be indicators of an inherited disorder. The pattern of inheritance in families is also informative. VWD is generally inherited in an autosomal dominant pattern (except for types 2N and 3, which are recessive), but can have variable penetrance and expressivity (particularly type 1). Factor deficiencies in FVIII and FIX are sex linked, and as such affected males present with more severe disease. Thus, the presentation of a male newborn with umbilical stump bleeding and a family history of hemophilia in a maternal uncle are strongly suggestive of an inherited factor deficiency. On the other hand, an elderly patient with no previous history of excessive bleeding despite surgical challenges who presents with recent onset of massive bruising and forearm compartment syndrome is much more consistent with an acquired bleeding diathesis. Diagnostic difficulties arise, however, when a mild familial disorder is present in a patient who previously has not been challenged because of young age, previous good health, or when the bleeding symptoms are highly variable within families, as can occur with mild disorders.
The positive family history may be informative, but a negative family history does not necessarily rule out a genetic disorder. Up to one-third of cases of hemophilia A and B arise as a result of new mutations, and the family history will be negative for abnormal bleeding. Because most new mutations causing hemophilia will manifest first in the carrier female,17,18 she may present with symptoms due to a decreased FVIII or FIX level without a diagnosis of hemophilia in the family. Alternatively, she may not have bleeding symptoms depending on her baseline factor level, which would vary by degree of random X chromosome inactivation (the Lyon hypothesis).19 Bleeding in patients with FXI deficiency occurs most commonly in those with severe deficiency, although severely affected individuals may be asymptomatic. Most heterozygotes are asymptomatic. However, the relation between the FXI level and bleeding is not clear-cut,20,21 and some heterozygotes bleed excessively after trauma. In patients where there is suspicion for an inherited bleeding disorder, gene mutation studies in family members may be helpful in confirming the diagnosis.
PHYSICAL EXAMINATION
The physical examination can provide helpful clues as to the etiology of the bleeding disorder, although many patients with mild bleeding disorders will have normal examinations. The skin, mucosa, and sites of skin compromise, such as vascular access sites or surgical or traumatic wounds, should be examined for ecchymoses, petechiae, oozing, and associated soft tissue hematomas. Palpable or larger ecchymoses (>3 cm), particularly in the absence of apparent trauma or on the trunk, are suggestive of an underlying disorder. Petechiae can be distinguished from small vasculitic, infectious, or embolic lesions in that they are never palpable, do not blanch, and tend to be distributed over the extremities, particularly the lower legs. Petechiae or mucosal purpura are seen in severe thrombocytopenia, but not usually in thrombocytopathies. On musculoskeletal examination, abnormal joint appearance and/or a loss of normal range of motion is suggestive of past hemarthroses, while active joint bleeding manifests with significant monoarticular arthralgia and effusion. Some rare disorders are associated with particular findings, such as abnormalities of the forearm in thrombocytopenia with absent radii, or defects in skin pigmentation in patients with syndromes associated with platelet granule or storage pool
disorders. The physical examination of all systems should be thorough in search of signs associated with systemic conditions that may predispose to bleeding, such as liver or kidney disease, malignancy, infection, autoimmune disease, or collagen-vascular disorders.
disorders. The physical examination of all systems should be thorough in search of signs associated with systemic conditions that may predispose to bleeding, such as liver or kidney disease, malignancy, infection, autoimmune disease, or collagen-vascular disorders.
LABORATORY EVALUATION
Careful history taking and clinical examination are essential components in the assessment of bleeding risk. The use of laboratory tests of coagulation and hemostasis cannot substitute for clinical assessment. Indeed, ample evidence exists that screening tests are unhelpful in the prediction of bleeding risk, especially when applied indiscriminately.22,23,24 Although there is much interest in developing a global assay of hemostasis, to date there is no single assay that can predict bleeding or thrombosis. The clinician must frequently use the results of several assays combined with the clinical presentation to arrive at a diagnosis or an assessment of bleeding risk. The primary use of coagulation testing should be to confirm the presence and type of bleeding disorder in a patient with a suspicious clinical history. An exception may be the preoperative assessment in infants and young children, in whom the clinical history is less likely to be informative.
Because of the nature of coagulation assays, proper sample acquisition and handling is critical to obtaining valid results. In patients with abnormal coagulation assays and no bleeding history, properly performed repeat studies are often normal. Most coagulation assays are performed in sodium citrateanticoagulated plasma that is recalcified for the assay. Because the anticoagulant is in liquid solution and needs to be added to blood in proportion to the plasma volume, incorrectly filled or inadequately mixed blood collection tubes will give erroneous results. Other patient factors can influence laboratory results. An elevated hematocrit (>55%) can result in an incorrect coagulation measurement because of a decreased plasma-to-anticoagulant ratio. An accurate result is obtained by adjusting the amount of sodium citrate solution added to the blood on the basis of the estimated plasma volume, using the formula:
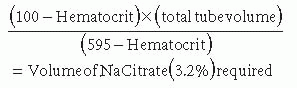
Complete Blood Count and Peripheral Smear
The complete blood count (CBC) is a necessary component of the evaluation of the bleeding patient and is often instrumental in identifying an underlying platelet defect or evidence of chronic or ongoing blood loss. However, artifacts are common, and verification by repeat or manual testing is warranted in patients with a newly identified CBC abnormality. Thrombocytopenia is often first identified by automated cell counting. An assessment of reticulated platelets or estimation of average platelet size from the mean platelet volume can also be useful clues to the underlying cause of thrombocytopenia. New diagnoses of thrombocytopenia should be verified by examination of the peripheral smear to evaluate the quantity and morphology of the platelets, to look for abnormalities in other blood cells, and to exclude pseudothrombocytopenia. A common cause of pseudothrombocytopenia is platelet clumping in the blood collection tube as a result of either the anticoagulant ethylenediaminetetraacetic acid (EDTA)-induced platelet clumping or specimen mishandling. Pseudothrombocytopenia may also result from the failure of the automated cell counter to identify morphologically abnormal platelets, as in the case of hereditary platelet disorders or acquired bone marrow diseases.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


