The last 4 decades have seen major advances in understanding the genetic basis of acute myeloid leukemia (AML), and substantial improvements in survival of children and young adults with the disease. A key step forward was the discovery that AML cells harbor recurring cytogenetic abnormalities. The identification of the genes involved in chromosomal rearrangements has provided insights into the regulation of normal hematopoiesis and how disruption of key transcription factors and epigenetic modulators promote leukemic transformation. Cytogenetics has been widely adopted to provide the framework for development of risk-stratified treatment approaches to patient management.
The last 4 decades have seen major advances in understanding the genetic basis of acute myeloid leukemia (AML), paralleled by substantial improvements in survival of children and younger adults with the disease. A key step forward was the discovery in the 1970s by pioneers in the field such as Rowley and colleagues that AML cells harbor recurring cytogenetic abnormalities, such as t(8;21)(q22;q22) in AML and t(15;17)(q22;q21) in acute promyelocytic leukemia, which was enabled by the introduction of chromosome banding techniques. This paved the way for the identification of the genes involved in chromosomal rearrangements, which has provided major insights into the regulation of normal hematopoiesis and how disruption of key transcription factors and epigenetic modulators promote leukemic transformation. To date, more than 100 balanced chromosomal rearrangements (translocations, insertions, and inversions) have been identified and cloned, with evidence suggesting that these are critical initiating events in the pathogenesis of AML. It has become apparent that karyotype analysis identifies biologically distinct subsets of AML that differ in their response to therapy and treatment outcome (reviewed in Refs. ). This information has been taken into account in the latest revision of the World Health Organization (WHO) classification, and cytogenetics has been widely adopted to provide the framework for development of risk-stratified treatment approaches to patient management.
Cytogenetic entities recognized in the WHO classification of AML
The latest (2008) revision of the WHO classification is organized in a hierarchical fashion, with the first group to be distinguished being AML with particular balanced translocations or inversions (and their molecular counterparts), defined as “AML with recurrent genetic abnormalities” ( Box 1 ), which includes the t(15;17)(q22;q21), the diagnostic hallmark of acute promyelocytic leukemia (APL), which is one of the common molecular subtypes of AML, accounting for ∼12% of cases. Cloning of the translocation breakpoints in the early 1990s showed that the t(15;17) leads to fusion of the gene encoding the myeloid transcription factor RARA (Retinoic Acid Receptor Alpha) with a previously unknown gene designated PML (ProMyelocytic Leukemia), which has subsequently been found to be involved in growth suppression and regulation of apoptosis (reviewed by Brown and colleagues ). Although most APL cases harbor an underlying PML – RARA fusion formed as a result of the t(15;17), often cytogenetically cryptic insertion events or more complex rearrangements (discussed later); in approximately 1% to 2% of APL cases, RARA is fused to an alternative partner. These latter cases are designated AML with a variant RARA translocation in the revised WHO classification, which include those involving ZBTB16 ( PLZF ), NPM1 , NUMA , FIP1L1 , and BCOR , formed as a result of the t(11;17)(q23;q21), t(5;17)(q35;q21), t(11;17)(q13;q21), t(4;17)(q12;q21), and t(X;17)(p11;q21), respectively, and PRKAR1A and STAT5B genes following rearrangements involving 17q. The nature of the fusion partner has an important bearing on disease biology, particularly the response to molecularly targeted therapies (ie, all- trans retinoic acid [ATRA] and arsenic trioxide [ATO]. Sensitivity to ATRA, which targets the ligand-binding domain in the RARα moiety in the C-terminal region of the fusion proteins (as well as wild-type RARα), has been documented in APL subtypes involving PML, NPM1, NUMA, and FIP1L1 ; whereas PLZF-RARα and STAT5B-RARα have both been associated with primary resistance to retinoids and a poorer prognosis. In the case of t(11;17)(q23;q21)/ PLZF – RARA –associated APL, the retinoid insensitivity is compounded by expression of the reciprocal RARα-PLZF fusion product, which functions as a transcriptional activator targeting PLZF-binding sites, leading to upregulation of cellular retinoic acid binding protein I (CRABP1), which sequesters retinoic acid, limiting its access to the nucleus. To date, sensitivity to ATO has only been shown in PML – RARA –positive APL, reflecting the capacity of arsenic to bind directly to the PML moiety of the fusion protein inducing its degradation via the proteosome. The importance of deregulation of retinoid signaling pathways in the pathogenesis of APL has been underlined by a recent report of a case with classic morphologic features, but with t(11;12)(p15;q13), leading to fusion of an alternative RAR family member (RARγ) with NUP98 , which is a recurrent target of chromosomal translocations in AML.
Cytogenetic abnormalities used to define entities within the WHO category of “AML with recurrent genetic abnormalities”
t(8;21)(q22;q22); RUNX1 – RUNX1T1
inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB – MYH11
t(15;17)(q22;q12); PML – RARA
t(9;11)(p22;q23); MLLT3 – MLL
t(6;9)(p23;q34); DEK – NUP214
inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1 – EVI1
t(1;22)(p13;q13); RBM15 – MKL1
Cytogenetic abnormalities sufficient to diagnose the WHO category of “AML with myelodysplasia-related changes”
Complex karyotype
(defined as 3 or more unrelated abnormalities, none of which can be a translocation or inversion associated with AML with recurrent genetic abnormalities)
Unbalanced abnormalities
−7 or del(7q)
−5 or del(5q)
i(17q) or t(17p)
−13 or del(13q)
del(11q)
del(12p) or t(12p)
del(9q)
idic(X)(q13)
Balanced abnormalities
t(11;16)(q23;p13.3) a
t(3;21)(q26.2;q22.1) a
t(1;3)(p36.3;q21.1)
t(2;11)(p21;q23) a
t(5;12)(q33;p12)
t(5;7)(q33;q11.2)
t(5;17)(q33;p13)
t(5;10)(q33;q21)
t(3;5)(q25;q34)
a A translocation commonly occurring in therapy-related AML. Before this translocation can be used as evidence for diagnosis of AML with myelodysplasia-related changes, therapy-related disease should be excluded.
Approximately 10% of AML are classified as having core binding factor (CBF) leukemia with balanced chromosomal rearrangements that disrupt genes encoding components of a heterodimeric transcription factor complex comprising RUNX1 (AML1, CBFα) and CBFβ, which plays a critical role in hematopoiesis. The CBFα subunit is targeted by the t(8;21)(q22;q22), which fuses RUNX1 ( AML1 , CBFA2 ) to the gene encoding the RUNX1T1 (formerly ETO [Eight Twenty-One]) transcriptional repressor, thereby potentially silencing RUNX1 target genes. The β subunit is targeted by the inv(16)(p13.1q22) or the less common t(16;16)(p13.1;q22), in which it is fused to the gene encoding myosin heavy chain ( MYH11 ). Although alternative fusion partners of CBFB have not been reported to date, RUNX1 is a recurrent translocation target in non-CBF acute leukemias, where it can be fused to ETV6 ( TEL ) as a result of the cytogenetically cryptic t(12;21)(p13;q22) in pediatric acute lymphoblastic leukemia (ALL), or to a range of partners in AML, including EVI1 / MDS1 as a result of t(3;21)(q26;q22) in “AML with myelodysplasia (MDS)-related changes” (see Box 1 ). RUNX1 has also been implicated in therapy-related leukemias arising following exposure to drugs targeting topoisomerase II.
The MLL (Myeloid/Lymphoid or Mixed-Lineage Leukemia) gene located at 11q23 is a further recurrent translocation target in acute leukemia, with more than 60 partner genes now characterized. MLL is an epigenetic regulator that plays a critical role in hematopoiesis, modulating HOX gene expression. The commonest MLL translocation observed in AML is the t(9;11)(p22;q23), which occurs in ∼2% of cases and leads to fusion of MLL with MLLT3 (formerly known as AF9 ). The t(9;11)(p22;q23), which has been associated with a relatively favorable outcome in some pediatric and adult AML studies, is distinguished as a separate entity in the latest WHO classification. Apart from the t(9;11), the most frequent other MLL translocations observed in AML are t(6;11)(q27;q23) involving MLLT4 ( AF6 ), t(11;19)(q23;p13.3) involving MLLT1 ( ENL ), t(11;19)(q23;p13.1) involving ELL , and complex rearrangements between 10p12 and 11q23 (eg, a reciprocal translocation and an inversion of an 11q segment translocated to 10p12 or inverted insertion of an 11q segment into 10p12 or a 10p segment into 11q23), in which the fusion partner is MLLT10 ( AF10 ). Although most translocations involving MLL occur in de novo leukemia, the locus is a recurrent translocation target in therapy-related leukemias, particularly those arising following exposure to the epipodophyllotoxin class of topoisomerase II poisons. Such cases are classified within the WHO as “therapy-related AML”.
The remaining subtypes of AML distinguished as separate disease entities by cytogenetics are those characterized by the t(6;9)(p23;q34), inv(3)(q21q26.2) or t(3;3)(q21;q26.2) and t(1;22)(p13;q13). The t(6;9)(p23;q34) is found in ∼1% of AML and leads to fusion of the DEK gene at 6p23 with NUP214 ( CAN ), which encodes a component of the nuclear pore complex. The inv(3) or t(3;3) occurs in a similar proportion of AML cases and is associated with upregulation of the zinc finger transcription factor EVI1, which is involved in normal hematopoiesis. AML with inv(3) or t(3;3) may present de novo, or secondary to prior MDS and is characterized by a normal or increased presenting platelet count and abnormal megakaryopoiesis with micromegakaryocytes in the marrow. EVI1 has long been implicated in leukemogenesis, having been identified as a recurrent integration target in murine retroviral mutagenesis screens ; further evidence has recently been provided by characterization of 2 cases of MDS associated with insertional activation of EVI1 occurring as a complication of gene therapy for chronic granulomatous disease. Analysis of these 2 cases provided important insights into potential mechanisms underlying acquisition of additional cytogenetic abnormalities in AML, with both cases showing loss of chromosome 7 (monosomy 7), which is well recognized as a frequent secondary abnormality in cases with inv(3) or t(3;3). Forced overexpression of EVI1 was found to disrupt normal centrosome duplication, suggesting that activation of EVI1 as a result of retroviral insertion or chromosomal translocation leads to genomic instability, giving rise to acquisition of additional changes such as monosomy 7 involved in progression to MDS and AML.
AML with t(1;22)(p13;q13) is extremely rare (to date, only ∼40 cases have been reported worldwide ) and is associated with acute megakaryoblastic leukemia occurring in infants and young children (<3 years of age), particularly those without Down syndrome. The translocation fuses RBM15 (for RNA-binding motif protein 15, also known as OTT ) with MKL1 (MegaKaryocyte Leukemia 1 or MAL ), which is involved in normal megakaryocyte maturation.
Apart from distinguishing AML with recurrent genetic abnormalities (as described earlier), for cases lacking one of these aberrations (or their molecular counterparts), cytogenetics has also been used in the latest WHO classification as a criterion (in conjunction with ≥20% leukemic blasts in the marrow) to define a subgroup of “AML with MDS-related changes”, as detailed in Box 1 . Some of these cytogenetic entities are common, such as monosomy 5 and 7 (−5 and −7) or deletion of the long arms of these chromosomes [del(5q) and del(7q)], which often occur as part of a complex karyotype. However, although −7 is recurrently observed both as a sole chromosome aberration and as part of a complex karyotype, −5 is rare in patients with noncomplex karyotypes. Moreover, most patients with a complex karyotype with −5 detected using banding techniques (eg, G-banding) also do not harbor true monosomy 5, because segments from a seemingly missing chromosome 5 can be found using spectral karyotyping or fluorescence in situ hybridization (FISH) in marker chromosomes or unbalanced structural aberrations only partially identified by G-banding. Although balanced translocations such as the t(11;16)(q23;p13.3), t(3;21)(q26.2;q22.1) involving MLL and RUNX1 , respectively, and the t(3;5)(q25;q34) that generates the NPM1 – MLF1 fusion may be initiating lesions in the development of leukemia, many of the changes considered as “MDS-related” entail loss of chromosomal segments or whole chromosomes, which are poorly understood at the molecular level and may represent secondary and cooperating lesions in AML pathogenesis (reviewed in Refs. ). Some of the aberrations designated MDS related, especially balanced translocations and idic(X)(q13), are extremely rare, making it challenging to characterize their biologic features and establish their impact on the clinical outcome.
Influence of patient age on cytogenetic profile in AML
AML is uncommon, with an overall incidence of approximately 4 cases per 100,000 population. Age has a major bearing on disease frequency and biology, with AML being particularly uncommon in children (0.6 cases/100,000), accounting for ∼20% of acute leukemias arising in this age group (<16 years). The disease increases in incidence in adults (2 cases/100,000 for ages 16–60 years), and is becoming an increasing health care problem as the population ages (incidence 9.7/100,000 for ages 60–74 years; incidence 20.2/100,000 for ages ≥75 years).
The pattern of cytogenetic abnormalities differs according to age. In infants, translocations involving the MLL gene account for a greater proportion of cases than in other age groups. In a series comprising 62 infants with AML (<1 year of age), 11q23 abnormalities were identified in 35%, with t(9;11) accounting for half of the cases. Apart from the t(1;22) associated with acute megakaryoblastic leukemia (described earlier), which is detected in ∼5% of infant AML, translocations involving the ETV6 ( TEL ) locus at 12p13 have also been particularly associated with AML presenting in this age group. The distinct pattern of cytogenetic abnormalities observed in AML presenting in infancy, compared with older children and adults, merits consideration for the potential reasons underlying this phenomenon and raises some interesting issues concerning the biology of these disorders. Many of the leukemias presenting in infancy develop in utero and hence the initiating mutations may arise in embryonic hematopoietic stem/progenitor cell populations, which may be distinct from the cellular origins of AML presenting in older children or in adulthood. The characteristics of the stem/progenitor cells, particularly in terms of nuclear architecture and the nature of the genes that are actively transcribed, may have a critical bearing on the chromosomal regions that are most susceptible to DNA damage and aberrant repair, which may provide an explanation for why rearrangements of MLL and ETV6 are particularly prevalent in both AML and ALL arising in children. Moreover, it is clear that fusion genes created by these translocations are extremely potent, giving rise to overt leukemia within short latency periods from occurrence of the initiating mutation.
In older children and younger adults, recurring balanced translocations/inversions, particularly t(15;17), t(8;21), and inv(16) or t(16;16), between them account for ∼30% of AML, whereas cytogenetic entities now defined as “MDS-related” in the WHO classification, particularly −5/del(5q), −7/del(7q), and complex karyotype, occur in ∼15% of cases. In older adults, there is a particular expansion of AML cases with chromosomal losses and gains, with an increase in cases with MDS-related changes including complex karyotype. This expansion mirrors the increased prevalence of secondary AML in older adults against the background of an antecedent hematological disorder, particularly MDS or myeloproliferative neoplasm, or occurring as a complication of treatment with chemotherapy and/or radiotherapy (which is an increasing problem as more patients survive their primary tumors).
Influence of patient age on cytogenetic profile in AML
AML is uncommon, with an overall incidence of approximately 4 cases per 100,000 population. Age has a major bearing on disease frequency and biology, with AML being particularly uncommon in children (0.6 cases/100,000), accounting for ∼20% of acute leukemias arising in this age group (<16 years). The disease increases in incidence in adults (2 cases/100,000 for ages 16–60 years), and is becoming an increasing health care problem as the population ages (incidence 9.7/100,000 for ages 60–74 years; incidence 20.2/100,000 for ages ≥75 years).
The pattern of cytogenetic abnormalities differs according to age. In infants, translocations involving the MLL gene account for a greater proportion of cases than in other age groups. In a series comprising 62 infants with AML (<1 year of age), 11q23 abnormalities were identified in 35%, with t(9;11) accounting for half of the cases. Apart from the t(1;22) associated with acute megakaryoblastic leukemia (described earlier), which is detected in ∼5% of infant AML, translocations involving the ETV6 ( TEL ) locus at 12p13 have also been particularly associated with AML presenting in this age group. The distinct pattern of cytogenetic abnormalities observed in AML presenting in infancy, compared with older children and adults, merits consideration for the potential reasons underlying this phenomenon and raises some interesting issues concerning the biology of these disorders. Many of the leukemias presenting in infancy develop in utero and hence the initiating mutations may arise in embryonic hematopoietic stem/progenitor cell populations, which may be distinct from the cellular origins of AML presenting in older children or in adulthood. The characteristics of the stem/progenitor cells, particularly in terms of nuclear architecture and the nature of the genes that are actively transcribed, may have a critical bearing on the chromosomal regions that are most susceptible to DNA damage and aberrant repair, which may provide an explanation for why rearrangements of MLL and ETV6 are particularly prevalent in both AML and ALL arising in children. Moreover, it is clear that fusion genes created by these translocations are extremely potent, giving rise to overt leukemia within short latency periods from occurrence of the initiating mutation.
In older children and younger adults, recurring balanced translocations/inversions, particularly t(15;17), t(8;21), and inv(16) or t(16;16), between them account for ∼30% of AML, whereas cytogenetic entities now defined as “MDS-related” in the WHO classification, particularly −5/del(5q), −7/del(7q), and complex karyotype, occur in ∼15% of cases. In older adults, there is a particular expansion of AML cases with chromosomal losses and gains, with an increase in cases with MDS-related changes including complex karyotype. This expansion mirrors the increased prevalence of secondary AML in older adults against the background of an antecedent hematological disorder, particularly MDS or myeloproliferative neoplasm, or occurring as a complication of treatment with chemotherapy and/or radiotherapy (which is an increasing problem as more patients survive their primary tumors).
Cytogenetics as a prognostic factor
Cytogenetics is the most powerful independent prognostic factor in AML and provides the framework for risk-stratification schemes that have been generally adopted to guide the treatment approach. Cytogenetic analysis is now considered a mandatory component of the diagnostic work-up of patients with suspected AML. Apart from identifying patients with particular subtypes of AML who may benefit from molecularly targeted therapies [ie, ATRA/ATO in t(15;17)/ PML – RARA -associated APL and tyrosine kinase inhibitors for patients with t(9;22)/ BCR – ABL1 -associated AML], karyotype analysis has been used to distinguish groups of patients with substantially different probabilities of achieving a complete remission (CR) and risks of relapse, particularly as a tool to guide transplantation in first CR. The major cooperative trial groups have typically distinguished between favorable, intermediate (standard), and adverse cytogenetic risk groups. However, several notable differences between the various classification systems have been described ( Table 1 ), which not only carries implications for patient management, but may also be a confounding factor when comparing trial results, particularly when drawing conclusions as to which groups of patients are most (and least) likely to benefit from allogeneic transplant in first CR. The t(15;17), t(8;21), and inv(16) or t(16;16) comprise the favorable risk group in most classification schemes; although some classifications do not include t(15;17), its favorable prognostic significance has been shown in numerous other studies. However, there has been a lack of consensus concerning the prognostic relevance of additional cytogenetic abnormalities in patients with CBF leukemia (see Table 1 ). In particular, the Southwest Oncology Group (SWOG) exclude from the favorable risk group those patients in whom the t(8;21) occurs as part of complex karyotype (defined as 3 or more abnormalities) and/or those in whom there is a deletion of the long arm of chromosome 9 [del(9q)], who account for 16% and 12% of patients with this subtype of CBF leukemia. The exclusion of the patients with del(9q) is based on an early, small study in which patients with this additional change were noted to have a poorer outcome. However, this finding has not been substantiated on analysis of much larger patient numbers ; complex karyotype has also not been found to adversely affect the treatment outcome of patients with t(8;21) [or those with inv(16) or t(16;16) ]. The SWOG group has also categorized del(16q) as favorable risk alongside the inv(16), although it is now clear that del(16q) cases are heterogeneous at the molecular level, with true del(16q) that are not misinterpreted inv(16) lacking an underlying CBFB – MYH11 fusion.
| Risk-group Assignment | Cytogenetic Abnormalities | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| MRC (Grimwade et al, 1998) | SWOG/ECOG a (Slovak et al, 2000) | CALGB b (Byrd et al, 2002) | GIMEMA/AML10 (Suciu et al, 2003) | GERMAN AMLCG (Schoch et al, 2003) | HOVON/SAKK (Cornelissen et al, 2007) | MRC Refined (Grimwade et al, 2010) | |||
| CR Rate | CIR | OS | |||||||
| Favorable | t(15;17), t(8;21), inv(16)/t(16;16) | t(15;17), t(8;21) [lacking del(9q) and complex karyotype], inv(16)/t(16;16)/ del(16q) | t(8;21), inv(16)/t(16;16) | t(8;21), inv(16)/ t(16;16) | t(8;21), inv(16)/t(16;16), del(9q) c | t(8;21), inv(16)/t(16;16) | t(15;17), t(8;21), inv(16)/ t(16;16) | t(8;21) with WBC <20×10 9 /L, inv(16)/t(16;16) and lacking adverse abn | t(15;17), t(8;21), inv(16)/t(16;16) |
| Intermediate | Normal karyotype, +8, +21, +22, del(7q), del(9q), abn(11q23), other structural/ numerical abn | Normal karyotype, +6, +8, −Y, del(12p) | Normal karyotype, −Y, del(5q), t(6;9), t(6;11), −7, loss of 7q, +8 sole, +8 with 1 other abn, del(9q), t(9;11), del(11q), +11, t(11;19)(q23;p13.1), +13, del(20q), +21 | Normal karyotype, −Y, t(9;11), del(9q), +8 sole, +8 with 1 other abn, +11, +13 | Normal karyotype, −Y, del(5q), loss of 7q, t(9;11), +11, del(11q), abn(12p), +13, del(20q), +21 | Normal karyotype, −Y | Normal karyotype, abn other than favorable or adverse | Normal karyotype, other noncomplex abn, pts without karyotype | Normal karyotype, abn other than favorable or adverse |
| Adverse | abn(3q) −5/del(5q) −7 complex karyotype (≥5 abn, excluding pts with favorable changes) | −5/del(5q), −7/del(7q), abn(3q), abn(9q), abn(11q), abn(20q), abn(21q), abn(17p), t(6;9), t(9;22), complex karyotype (≥3 abn) | inv(3) or t(3;3), abn(12p), complex karyotype [≥3 abn, excluding pts with t(8;21), inv(16)/t(16;16) and t(9;11)] | −7, +21, complex karyotype [≥3 abn, excluding pts with t(8;21), inv(16)/t(16;16) and t(9;11)] | inv(3) or t(3;3), t(6;9), t(6;11), −7, +8 sole, +8 with 1 other abn, t(11;19) (q23;p13.1), complex karyotype [≥3 abn, excluding pts with t(8;21), inv(16)/t(16;16) and t(9;11)] | complex karyotype (unspecified), abn(3q), −5/del(5q), −7/del(7q), t(6;9), t(9;22), abn(11q23), absence of favorable abn d | inv(3)/t(3;3) −5/del(5q) −7/del(7q) abn(11q23) del(12p) abn(17p) complex karyotype (≥3 abn) | abn(3q) −5/del(5q) −7/del(7q) abn(11q23) t(6;9) t(9;22) complex karyotype (≥3 abn), pts with intermediate cytogenetics with a late CR, reached after cycle II of induction | abn(3q) [excluding t(3;5)], inv(3)/t(3;3), add(5q)/del(5q)/−5, add(7q)/del(7q)/−7 (excluding pts with favorable karyotype), t(6;11), t(10;11), t(11q23) [excluding t(9;11) and t(11;19)], t(9;22), −17/abn(17p), complex karyotype (≥4 abn, excluding pts with favorable or adverse changes) |
a All other, unclassified abnormalities were considered to have unknown risk.
b Abnormalities not specified as conferring favorable, intermediate, or adverse risk were not included in the risk assessment model.
c Favorable for a group of 13 patients with del(9q) that included 6 who underwent transplantation off protocol; intermediate for nontransplanted patients treated with chemotherapy only.
d The abnormalities listed were denoted as “very bad.” This classification also included “bad” cytogenetics, which was defined as “the presence of other abnormalities without good or very bad cytogenetic features.”
A normal karyotype is identified in ∼40% to 45% of AML cases presenting in adults aged 16 to 60 years and, overall, these patients have an intermediate prognosis. However, as discussed later and elsewhere, cytogenetically normal AML (CN-AML) is highly heterogeneous at the molecular level, and mutation screening is now widely applied to refine risk stratification in this group (see later discussion). Several cytogenetic abnormalities have been universally considered to confer a poor prognosis [eg, −5/del(5q), −7, and inv(3) or t(3;3)]. However, there has been little consensus as to the outcome of various structural and numerical abnormalities, including cases with rare recurring cytogenetic abnormalities (ie, with individual incidence <2%), which together account for approximately 10% of AML and have variably been considered to predict an intermediate or adverse prognosis (see Table 1 ). Another major source of variability is in the definition of a complex karyotype, with most groups applying this term to karyotypes comprising at least 3 unrelated abnormalities [in most instances excluding t(15;17), t(8;21), and inv(16) or t(16;16)], whereas in the original UK Medical Research Council classification, a higher cutoff of greater than or equal to 5 abnormalities was adopted but later revised to greater than or equal to 4 abnormalities.
There are significant differences in the risk-group classifications of some of the rarer cytogenetic entities, which could reflect differences in the age profiles of the patients studied, as well as problems in obtaining reliable outcome data because of small sample sizes. However, it is also possible that interstudy and intrastudy differences in treatment could have played a part, affecting the outcome of particular cytogenetically defined subsets of AML.
Despite the challenges, it would be helpful if greater standardization in risk stratification in AML could be achieved, to facilitate development of optimized treatment approaches and reporting of outcome data, thereby enabling more reliable comparison of results from different international trial groups. Establishing robust outcome data for particular cytogenetic subgroups is clinically important, taking into account the results of a recent meta-analysis that has suggested that a relapse risk in excess of 35% can provide a useful working threshold to identify patients in whom allogeneic transplant may confer a survival benefit. Therefore, patients with favorable risk AML [ie, those with t(15;17)/ PML – RARA and CBF leukemia with t(8;21)/ RUNX1 – RUNX1T1 or inv(16)/t(16;16)/ CBFB – MYH11 ] are no longer routinely subject to transplantation in first CR, which confers no overall survival advantage.
Refinement of Cytogenetic Classification in Younger Adults with AML
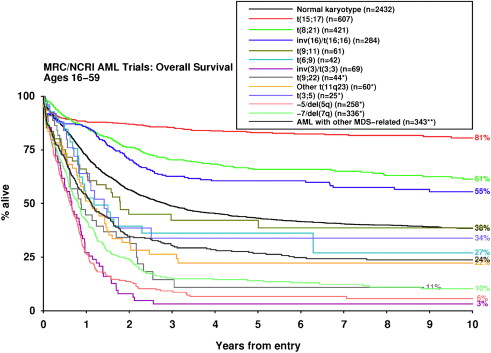
In an attempt to reconcile differences between several existing cytogenetic classification systems used to direct therapy in younger adults, the UK Medical Research Council (MRC) examined outcomes for a large number (>50) of cytogenetic entities within a cohort of 5876 patients (median age 44 years) who received comparable therapy in successive clinical trials, with prolonged follow-up (median 7.3 years). In multivariable analysis taking into account age, presenting white blood cell count (WBC) and type of AML (de novo vs secondary), the t(15;17), t(8;21), and inv(16) or t(16;16) were robustly confirmed as powerful independent predictors of favorable outcome, irrespective of the presence of additional cytogenetic abnormalities ( P <10 −12 ). In patients with inv(16), the presence of additional abnormalities, particularly trisomy 22, was associated with a significantly better outcome, in accordance with previous studies. This finding may relate to differences in presenting WBC, which is significantly higher in patients with inv(16) as the sole abnormality and cannot be explained by differences in the frequency of KIT mutations, which constitute an adverse prognostic factor in patients with inv(16) or t(16;16). The multivariable analysis conducted by the MRC group did not reveal any novel cytogenetic entities conferring a favorable outcome. However, a large number of abnormalities that independently predicted an adverse prognosis were identified (see Table 1 ). Apart from −5/del(5q), −7/del(7q), and inv(3) or t(3;3), which are well recognized as conferring adverse risk, the study also confirmed that t(9;22)(q34;q11), which had hitherto been the subject of small case series, also predicts a poor prognosis ( Fig. 1 ). There have been conflicting data for the outcomes of patients with translocations involving 11q23, which have been considered to confer a poor prognosis, with the possible exception of those with the t(9;11). In the MRC series, better outcome was observed in patients with the t(9;11)(p22;q23) and t(11;19)(q23;p13), although involvement of MLLT1 and ELL was not distinguished; whereas patients with t(6;11)(q27;q23) and t(10;11)(p12;q23) involving MLLT4 and MLLT10 , respectively, or other 11q23 abnormalities were found to have a significantly poorer outcome in multivariable analysis. Although the t(6;9)(p23;q34) is widely considered to predict a poor outcome in AML, which may in part reflect its frequent association with internal tandem duplication (ITD) in the juxtamembrane domain of the FLT3 gene encoding Fms like tyrosine kinase 3 ( FLT3 -ITD), and was also associated with a high relapse rate in the MRC series (62% at 10 years), the effect was not sufficiently strong to emerge in the multivariable analysis. The MRC study also sought to establish the most robust cutoff to define a karyotype as complex. For this analysis, cytogenetic entities that, in their own right, conferred a favorable or adverse prognosis in the multivariable analysis were excluded, and the impact of complexity was considered solely in the context of standard risk cytogenetic abnormalities. In contrast with previous studies that used cutoffs of 3 or 5 abnormalities to define a complex karyotype, the MRC study suggests that the presence of at least 4 unrelated abnormalities provides the most appropriate cutoff, defining a group with significantly poorer prognosis.
The Dutch HOVON (Haemato Oncology Foundation for Adults in the Netherlands) group also recently revisited the cytogenetic classification of AML and distinguished a subgroup of patients with a so-called monosomal karyotype (MK) that is characterized by a particularly poor prognosis. Patients with non-CBF AML were defined as having a monosomal karyotype from the loss of an autosomal chromosome (ie, excluding −Y and −X) in combination with at least 1 other autosomal monosomy (eg, 44,XY,−7,−18) or 1 or more structural abnormality (eg, 45,XY,inv(3)(q21q26),−7). Monosomal karyotypes almost invariably include at least 1 chromosomal abnormality that would independently be associated with adverse risk. Moreover, because up to 75% of complex karyotypes are hypodiploid (ie, have ≤45 chromosomes) and several pseudodiploid (46 chromosomes) and hyperdiploid complex karyotypes (≥47 chromosomes) also include at least 1 monosomy, most patients with a complex karyotype are considered to have a monosomal karyotype. Overall, approximately half of patients defined as having an adverse karyotype according to standard classification systems have a monosomal karyotype, and several studies have confirmed that the subgroup with MK+ AML have a particularly poor prognosis. However, there are preliminary data to suggest that their outcome could be improved by allogeneic transplantation in first remission.
Prognostic Significance of Cytogenetics in Pediatric AML
There have been few large studies considering the impact of cytogenetics on outcome in pediatric AML. The Pediatric Oncology Group (POG) considered a cohort of 478 patients aged less than 21 years, reporting the best outcomes in those with inv(16), t(8;21) and normal karyotype. Patients with the t(15;17), most of whom did not receive ATRA, and those with various 11q23 abnormalities had a poor prognosis. The findings of a more recent, similarly sized study (454 patients <18 years of age) published by the Berlin-Frankfurt-Műnster (BFM) group were in accordance with most adult series, showing the best outcomes in patients with inv(16), t(8;21), and t(15;17). Significantly poorer outcomes were observed in patients with monosomy 7 and 11q23 aberrations [apart from t(9;11) as the sole abnormality and the t(11;19)]. The largest study to date was conducted by the UK MRC group and considered the prognostic significance of 22 cytogenetically defined subgroups within a cohort of 729 children aged 0 to 15 years. In this study, in which patients with APL were excluded, the best outcomes were observed in the CBF AML, with 10-year overall survivals of ∼80%. In contrast with the POG and BFM studies, patients with 11q23 abnormalities (n = 104) had an intermediate prognosis (61% OS at 10 years), with no evidence of heterogeneity according to the translocation partner. Although this study did not analyze the prognostic significance of secondary aberrations accompanying 11q23 abnormalities, a recent large international effort identified trisomy 8 as independently favorable, and trisomy 19 as adverse, prognostic factors in pediatric 11q23/ MLL -rearranged AML, whereas complex karyotype was found to be an unfavorable prognosticator in univariable analysis only. In a series analyzed by Harrison and colleagues, several abnormalities that have been associated with adverse risk in adult patients (eg, −5, 3q abnormalities and complex karyotype) were not predictive of a worse outcome. However, significantly poorer survivals were observed in pediatric AML with 5q abnormalities, t(6;9), monosomy 7, trisomy 13, and 12p abnormalities, including translocations involving ETV6 , with 36% of patients alive at 10 years.
Prognostic Significance of Cytogenetics in Older Adults with AML
The outcome of AML presenting in older adults is poorer (7%–15% 5-year survival for adults >60 years old), compared with when the disease presents in younger individuals. Nevertheless, cytogenetics are still predictive in this age group. The largest study to date was conducted by the MRC group, studying 1065 patients (aged >55 years) treated in the AML11 trial. The best outcome was observed in patients with APL with the t(15;17) and those with CBF leukemia, with a superior CR rate (72%) associated with low rates of resistant disease. However, overall survival rates were poorer than those observed in younger adults with the same cytogenetic abnormalities (only 34% OS at 5 years). Although the relapse rate in patients with APL with the t(15;17) was low (26% at 5 years), the relapse rates in the patients with CBF leukemia were substantially higher (>80% relapse risk at 5 years) compared with younger patients, which may reflect differences in disease biology as well being a consequence of less intensive therapy. Older patients with a complex karyotype (defined as 5 or more abnormalities) had an extremely poor prognosis, with few achieving CR (26%) because of high rates of resistant disease (56%). The small proportion of elderly patients achieving remission almost invariably relapsed, leading to overall survival rates of only 2% at 5 years. Similar data have been published by other groups, raising the question of whether elderly patients with high-risk cytogenetics might be most appropriately offered palliative nonintensive treatment approaches or supportive care. In contrast, recent preliminary data indicate that at least some patients aged 60 to 70 years who achieve a CR may benefit from reduced-intensity allogeneic transplantation. Moreover, alternative, novel therapies are also being investigated in older adults with AML. In the MRC series, patients with normal karyotypes or noncomplex cytogenetic aberrations had marginally better outcomes, with 5-year survivals of 15% and 10%, respectively. It is likely that the slightly better outcome in CN-AML occurring in older patients may reflect the influence of cases with NPM1 gene mutation (see later discussion). The Cancer and Leukemia Group B (CALGB) and German AML Study Groups have both reported on the prognostic impact of cytogenetics in older adults, also finding evidence for a better outcome in patients with the t(15;17) and CBF leukemia, and similarly reporting that patients with a complex karyotype have a dismal prognosis. Most recently, the GOELAMS (Groupe Ouest Est d’Etude des Leucémies et Autres Maladies du Sang) group showed in a study involving 186 older adults (>60 years) with unfavorable cytogenetic abnormalities that presence of a monosomal karyotype (found in 59%) predicted a particularly poor prognosis.
Related posts:
 Acute Leukemias
Acute Leukemias
 Clinical Implications of Novel Mutations in Epigenetic Modifiers in AML
Cellular Therapies in Acute Lymphoblastic Leukemia
Clinical Implications of Novel Mutations in Epigenetic Modifiers in AML
Cellular Therapies in Acute Lymphoblastic Leukemia
 Novel Transplant Strategies in Adults with Acute Leukemia
Novel Transplant Strategies in Adults with Acute Leukemia
 Prognostic Factors in Adult Acute Leukemia
Prognostic Factors in Adult Acute Leukemia
 Induction and Postremission Strategies in Acute Myeloid Leukemia: State of the Art and Future Directions
Induction and Postremission Strategies in Acute Myeloid Leukemia: State of the Art and Future Directions
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


