Development of the Vasculature, Angiogenesis, and Lymphangiogenesis
Aernout Luttun
Peter Carmeliet
As species have become larger and more complex during evolution, nature has provided them with two tubular transport and communication systems, the blood vascular and the lymphatic system. While the former is responsible for oxygen and nutrient delivery to nearly all tissues, the latter mostly runs alongside the blood vascular system and serves to recirculate excess interstitial fluid (ISF) to take up lipids from the gastrointestinal tract and to regulate immune responses. The blood vascular system forms a closed loop with three main segments: the arteries carrying oxygen-rich blood from the heart, the capillaries that exchange oxygen with the tissues, and the veins that bring the deoxygenated blood back to the heart. In contrast, the lymphatic system is a blind-ended circuit beginning with the initial lymphatics (capillaries; specialized in fluid uptake) that connect to collector vessels (which pump the lymph fluid forward) that end up in larger ducts. These ducts (the thoracic duct and the right lymphatic duct) connect the lymphatic system with the venous arm of the blood vascular system. Given their ubiquitous presence in nearly all organs of the body, both vascular systems are frequent targets of disease. For instance, excessive blood vessel growth promotes cancer growth, while a shortage of blood vessels leads to ischemia and ultimately tissue death. Impaired lymph clearance results in lymphedema, characterized by disfiguring tissue swelling, while increased lymphatic growth can result in metastatic spreading of tumors. Hence, in order to remedy these diseases, a thorough understanding of the development of both vascular systems is paramount.
Both systems are lined on the inside (“endo” in Greek) by endothelial cells (ECs). While this monolayer of cells was long thought to be merely a passive barrier between tissues and blood or lymph, now we have come to understand that ECs are dynamic sensors that continuously adapt their behavior to their environment. This versatile behavior results in remarkable morphologic, functional, and molecular differences, a phenomenon known as “endothelial heterogeneity.”1,2,3 We distinguish different levels of heterogeneity. During vascular growth, the EC can adopt at least three distinct functional behaviors, that is, the tip, stalk, or phalanx cell. In addition, “macrovascular heterogeneity” refers to the differences between ECs from arteries, veins, and lymphatic vessels, while “microvascular heterogeneity” relates to the differences at capillary level in distinct organs. The existence of EC heterogeneity has vast consequences for disease development and hence should be taken into account when designing new therapies based on stimulating or inhibiting vessel growth. While proper blood/lymphatic vessel formation also involves non-EC types (e.g., smooth muscle cells or SMCs and hematopoietic cells), in this overview we describe the development of both vascular systems mainly from an endothelial perspective.
THE BLOOD VASCULAR SYSTEM
On the Origin of Blood Vascular Endothelium
Endothelia in the developing vascular system have a common origin, the mesoderm. The mesoderm is one of the three embryonic germ layers that segregate during gastrulation, starting around embryonic day (E) 6.25 in the mouse embryo (reviewed in Refs.4,5). At this time, cells from the epiblast converge toward the posterior proximal pole of the embryo to form the primitive streak (PS), which subsequently extends in the anterior direction to the distal tip of the embryo (reviewed in Ref.6). Mesoderm is formed when epiblast cells entering the streak undergo epithelial-to-mesenchymal transition (EMT). Distinct mesodermal zones become patterned according to the time and site of passage through the PS (figure 38.1; reviewed in Refs.4,7): the earliest, most posterior cells become extraembryonic mesoderm (the progenitor of yolk sac blood islands), the intermediate somewhat later invading cells become lateral plate (giving rise to the intraembryonic axial vessels), paraxial (giving rise to the somites), and cardiac mesoderm and the epiblast cells that enter through the anterior tip of the streak become axial mesoderm (comprising the notochord, the prechordal plate, and the node). Interestingly, each of these mesoderm layers has the intrinsic capacity to form ECs, with the exception of the axial mesoderm.8
Mesoderm induction, EMT, and mesoderm patterning involve a complex interplay and gradient of signals (including bone morphogenetic proteins [BMPs], Wnt, fibroblast growth factors [FGFs], nodal, and Hox proteins).4,5,6,9,10 As epiblast cells have to move extensively during PS formation and mesoderm patterning, they do so by following certain physical (e.g., a basal lamina underneath the moving cells) and chemical guidance cues. In the avian embryo, this involves attractive and repulsive signals (e.g., SDF-1/CXCR4, cAMP, FGFs/FGFR; Refs.4,6). While moving, epiblast cells extend lamellipodia/filopodia in the direction of migration.6 The idea that this process may resemble the movement of angiogenic sprouts or neural growth cones, which involves certain classes of guidance molecules (see below), is intriguing. For instance, the EphB4 receptor (expressed together with its ligand ephrinB2 in embryoid bodies) was recently shown to be involved in modulation of mesoderm induction, which may imply a role in migration/patterning as well.11 Regarding the location of the emergence of ECs, two main regions can be distinguished, corresponding to two temporal waves of vascular development, extraembryonic
and intraembryonic, the former starting with the migration of EC precursors into the yolk sac (around E6.5 to 7.0 in the mouse12) and the latter starting slightly later around E7.3 with the appearance of EC precursors in the cranial region.13
and intraembryonic, the former starting with the migration of EC precursors into the yolk sac (around E6.5 to 7.0 in the mouse12) and the latter starting slightly later around E7.3 with the appearance of EC precursors in the cranial region.13
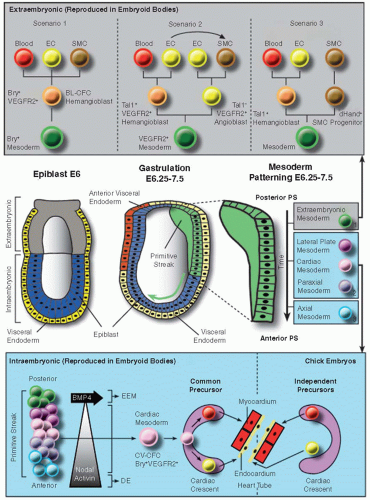 FIGURE 38.1 On the origin of blood vascular ECs. All embryonic tissues derive from the epiblast (blue in left middle panel). During gastrulation (central middle panel), a subset of epiblast cells converges toward the posterior proximal pole to generate the primitive streak (PS; green structure in right and central middle panel) that expands toward the anterior pole (green arrow). Depending on timing (indicated by a time axis and numbering in boxes on the right) and location (across the anterior-posterior PS axis) of passage through the PS, epiblast cells give rise to distinct mesodermal derivatives. Extraembryonic hematopoiesis and vasculogenesis can be reproduced in a culture system based on ESCs, that is, embryoid bodies. At least three scenarios (gray box at the top) have been described for the cellular hierarchy that gives rise to the first hematopoietic and vascular precursors. In scenario 1, hematopoietic cells, endothelial cells (ECs), and smooth muscle cells (SMCs) arise from a common precursor, “the hemangioblast” (its corresponding ESC-derived culture equivalent is called “blast colony-forming cell” or BL-CFC). According to scenario 2, SMCs and blood cells derive from separate precursors; however, hemangioblast-derived ECs may transdifferentiate to form SMCs (indicated by the curved arrow). In scenario 3, SMCs arise from a dHand+ unipotent precursor. In the lower blue box, one example is shown of how the embryoid body system has been used to further reveal cell hierarchies during the formation of intraembryonic mesoderm derivatives. Passage through the PS can be mimicked by exposing the cultures to different levels of activin/nodal and BMP-4 expression. Intermediate activin/nodal concentrations result in the formation of a common cardiogenic mesoderm precursor, the cardiovascular colony-forming unit or “CV-CFC,” giving rise in the cardiac crescent to (endocardial) ECs, cardiomyocytes (together forming the primitive heart tube), and SMCs (not shown). Studies in chick embryos have suggested an alternative scenario where endocardial and myocardial lineages derive from independent precursors in/outside the cardiac crescent. Bry, brachyury; VEGFR2, vascular endothelial growth factor receptor-2. (Adapted from Kattman SJ, Adler ED, Keller GM. Specification of multipotential cardiovascular progenitor cells during embryonic stem cell differentiation and embryonic development. Trends Cardiovasc Med 2007;17(7):240-246; Harris IS, Black BL. Development of the endocardium. Pediatr Cardiol 2010;31(3):391-399.) |
Emergence of Extraembryonic EC Precursors
The extraembryonic mesoderm in birds and mammals is organized in two layers separated by the coelom, the somatopleural mesoderm (adjacent to ectoderm), and the splanchnopleural mesoderm (aligned with endoderm). EC emergence in the splanchnopleural mesoderm of the yolk sac occurs simultaneously with the appearance of primitive erythrocytes. This has fueled the idea of the existence of a common precursor, “the hemangioblast,” a term that was already coined in 1932 by Murray.14 In vitro evidence for the hemangioblast was given by the discovery of a bipotential precursor, called the “blast colony-forming cell” (BL-CFC) during mouse and human embryonic stem cell (ESC) differentiation, a system that faithfully recapitulates the early stages of vasculogenesis and (primitive) hematopoiesis (reviewed in Refs.15,16). In vivo evidence for the hemangioblast was shown in reporter mice expressing GFP under the brachyury (Bry) promoter12 and in zebrafish.17 Interestingly, the hemangioblast concept is evolutionary conserved since a similar vascular/hematopoietic precursor has been described in Drosophila melanogaster.18 Around E6.5 to 7.0 in the mouse, the Bry-GFP+ precursors, coexpressing vascular endothelial growth factor receptor-2 (VEGFR2), emerge in the posterior region of the PS and move into the yolk sac. There, they form polarized aggregates in which the outer cells give rise to flat ECs and the inner cells become primitive erythrocytes. Several of these structures subsequently fuse to form a rudimentary capillary network.7,19
Not all yolk sac blood vessels arise from hemangioblasts. Initial static observations of stained mouse embryos suggesting that some unipotential “angioblasts” are present in the yolk sac13 were later confirmed by kinetic analysis of mouse embryonic/stromal cell cocultures (a system equivalent to the early gastrulating embryo), where two waves of EC precursors were found, one consisting of angioblasts and a second composed of hemangioblasts.20 The relative importance of angioblast versus hemangioblast contribution to ECs is still a matter of debate since clonal analysis revealed that only a minor portion of ECs derives from hemangioblasts.21 In addition to ECs and blood cells, the yolk sac mesoderm also harbors SMCs. Several scenarios exist concerning their origin (FIGURE 38.1). One theory claims that a common Bry+VEGFR2+ precursor (the hemangioblast) exists for the three lineages (blood, EC, and SMC).12,15,22 According to another study, a VEGFR2+ mesodermal precursor first segregates into a VEGFR2+Tal1+ hemangioblast (which can give rise to hematopoietic stem cells or HSCs and ECs) and a VEGFR2+Tal1– angioblast (which can give rise to ECs and SMCs). A similar VEGFR2+ progenitor with EC and SMC potential was described earlier by Yamashita et al.23 This second model also considers the possibility that SMCs can arise by transdifferentiation from ECs, which would mean the hemangioblast can also be a common source for the three lineages.24 This transdifferentiation may however be a culture-restricted phenomenon that does not occur in vivo.25 A third possibility was recently proposed in the avian embryo, where the extraembryonic mesoderm would give rise to a Tal1+ bipotential endothelial/hematopoietic precursor (the hemangioblast) and a separate dHand+ SMC precursor, the segregation of which would be positively regulated by Notch signaling in cooperation with Wnt signaling.26 A stimulatory effect of Notch on SMC differentiation at the expense of hematopoietic/endothelial fate was also shown during mouse ESC differentiation.27 Finally, the transient nature of the hemangioblast has made it difficult to prove its existence during development; however there is now also evidence to suggest that such a bipotential progenitor may also persist during adulthood.28,29
Emergence of Intraembryonic EC Precursors
The first intraembryonic VEGFR2+ EC precursors emerge in the cranial region of the mouse embryo around E7.3 where they contribute to the endocardium, the EC monolayer lining the primitive heart tube. A little thereafter (at E7.8), the VEGFR2+ primordia of the bilateral dorsal aorta (DA) arise, followed by the lateral vascular networks (between E8 and E8.5) (see below). It has been suggested that the vast majority of ECs within the embryo arises from angioblasts with limited contribution of hemangioblasts.30 In zebrafish, hemangioblast-derived ECs all clustered in a specific posterior region of the axial vessels17 but were later also found more anteriorly.31,32 Hemangioblast contribution to ECs has also been suggested in other regions, for example, the heart (see below). In agreement with hemangioblast activity around the axial vessels in zebrafish, in the mammalian embryo, the floor of the DA in the aorta-gonadmesonephros region was shown to contain HSCs in intimate contact with the endothelial wall.
Two hypotheses exist concerning the relationship of the ECs and HSCs at this location.16,33,34 According to one view, HSCs
may arise independent of the ECs from a mesodermal precursor in a region underneath the aortic endothelium, called “the subaortic patches” (only seen in mammals, not in lower vertebrates), and then subsequently move across the DA floor into the lumen. However, recent studies have favored another scenario that HSCs arise within the DA lumen as descendants of cells with endothelial characteristics, called “the hemogenic endothelium.” Recent in vivo tracing experiments as well as ESC cultures have strongly supported such a process.35,36,37,38 Interestingly, in birds, the aorta has been shown to be a chimeric structure, with ECs of the floor originating from the splanchnopleural mesoderm (also called the lateral plate mesoderm or LPM) and the roof deriving from the paraxial/somitic mesoderm (see below). In a later stage, the floor ECs are replaced by ECs coming from the somites, and this transition coincides with cessation of hematopoietic activity.16,25,33 Since the paraxial mesoderm and LPM derive from distinct regions and at distinct time points during mesodermal patterning in the PS (see above), it is possible that the different hematopoietic capacities are already determined at that level.
may arise independent of the ECs from a mesodermal precursor in a region underneath the aortic endothelium, called “the subaortic patches” (only seen in mammals, not in lower vertebrates), and then subsequently move across the DA floor into the lumen. However, recent studies have favored another scenario that HSCs arise within the DA lumen as descendants of cells with endothelial characteristics, called “the hemogenic endothelium.” Recent in vivo tracing experiments as well as ESC cultures have strongly supported such a process.35,36,37,38 Interestingly, in birds, the aorta has been shown to be a chimeric structure, with ECs of the floor originating from the splanchnopleural mesoderm (also called the lateral plate mesoderm or LPM) and the roof deriving from the paraxial/somitic mesoderm (see below). In a later stage, the floor ECs are replaced by ECs coming from the somites, and this transition coincides with cessation of hematopoietic activity.16,25,33 Since the paraxial mesoderm and LPM derive from distinct regions and at distinct time points during mesodermal patterning in the PS (see above), it is possible that the different hematopoietic capacities are already determined at that level.
While the ESC system was originally thought to represent the early, mostly extraembryonic stages of vasculogenesis/(primitive) hematopoiesis, recent advances have made it possible to adapt the system to also study several aspects of intraembryonic mesodermal fate mapping.15,16,22 Based on the expression of an anterior PS marker, Foxa2, anterior (Bry+Foxa2high; induced by activin/nodal and giving rise to cardiogenic mesoderm) and posterior (Bry+Foxa2low/-; induced by Wnt signaling and giving rise to extraembryonic mesoderm) PS could be segregated.39 Another group also demonstrated that different mesoderm types can be segregated in ESC cultures using specific tracers and culture conditions and that the common or single potential to make ECs and blood was different among these types.40 Using their murine and human ESC differentiation system, Keller’s group showed that in addition to the Bry+VEGFR2+ or BL-CFC population that is the in vitro equivalent of the hemangioblasts colonizing the yolk sac and originating from the posterior PS, a second Bry+VEGFR2+(Islet1+) population, the “cardiovascular colony-forming cell” (CV-CFC; FIGURE 38.1), can be obtained that is representative of the cardiogenic mesoderm precursor that originates from the anterior PS and gives rise to (endocardial) ECs, cardiomyocytes, and SMCs in vitro.22 Others have confirmed that in embryoid bodies, these three lineages can arise from a common VEGFR2+(Islet1+) precursor.41 While these ESC-based studies have thus suggested a common origin for endocardial and myocardial lineages, another model of endocardial specification based on lineage tracing by cell labeling in chick and zebrafish embryos suggests that the endocardial lineage develops from an independent precursor in the cardiac crescent—the structure that gives rise to the primitive heart tube consisting of an endocard and myocard layer (both conceptual models have been reviewed in Ref.42; FIGURE 38.1). A recent study using fluorescent mapping/time-lapse imaging and quail tissue/mouse EC implantation in chick embryos supports the view that endocardium at least in part derives from endothelial-restricted precursors devoid of myocardial differentiation potential and which may reside outside the cardiac crescent (FIGURE 38.1).43
Vasculogenesis
Once EC precursors have emerged from their mesodermal origin, they need to acquire a more differentiated EC signature and be assembled and shaped into a primitive and disperse tubular network to lay down the basic architecture of the embryonic vasculature. This process is collectively called “vasculogenesis.” Much of this happens before the first heartbeat, while further reshaping occurs after the onset of blood flow. The molecules that orchestrate this complex process are only partially known. Nevertheless, VEGF-A most likely is the conductor of this orchestra since it is expressed in spatial and temporal association with most—if not all—events during vascular formation. Indeed, lack of a single VEGF allele disrupts embryonic vascular patterning.44,45 Complementary to this, the VEGFR2 receptor is the earliest marker to define the endothelial lineage, and cells subsequently express additional markers such as junctional molecules (VE-cadherin and CD31), CD34, and later Tie2.13 This temporal sequence of expression is more or less recapitulated during EC differentiation of mouse ESCs.46 In the mouse, the first VEGFR2+ EC precursors are spotted at E7.3 in the cranial region corresponding to the endocardial primordia, while precursors that will give rise to the bilateral DA are first detectable around E7.6 flanking the notochord.13 The region around the notochord remains avascular at this stage due to notochord-derived negative signals, that is, BMP antagonists chordin and noggin.47 Subsequently, these DA EC precursors gradually merge into a tubular structure in a bidirectional fashion along the craniocaudal axis. Additionally, between E8 and E8.5, primary vascular networks arise laterally from this axis, called the “lateral vascular networks,” that connect to the extraembryonic vasculature.13 The first axial vein (the cardinal vein) emerges slightly later than the DA. In the following paragraphs, we give an overview of the different steps in vasculogenesis during development, thereby using specific examples and high-lighting some recently identified novel concepts.
Although the extraembryonic tissues also go through a vasculogenic phase slightly earlier than within the embryo,7 here we describe the different steps of the process using the sculpture of the first intraembryonic main axial vessels, the DA, and the posterior cardinal vein (PCV), as an example (FIGURE 38.2). Axial vessel formation has been studied in various species, including quail embryos, chick embryos, (transgenic) mouse fetuses, zebrafish embryos, and Xenopus tadpoles. There are some important differences between these species. In birds and mammals, the DA is initially paired and needs to merge into a single tube. In amphibians and fish, DA patterning is in part driven by the hypochord, a temporal tubular structure that does not exist in birds and mammals and lies just ventral from the notochord.48 As such, there are slight variations in the mechanisms by which the axial vessels are formed (FIGURE 38.2). A first critical step in DA and PCV formation is migration of the EC precursors to the correct location, that is, the midline of the embryo ventral from the notochord. The process involves a combination of soluble factors, cell-cell interactions and cell-extracellular matrix interactions.49 In zebrafish and Xenopus, axial vessel EC precursors originate and migrate mainly—if not exclusively— from the LPM.50,51 The underlying endoderm and the overlying midline structures (notochord/hypochord) seem to direct this migration. In Xenopus, expression of the diffusible VEGF122 isoform in the hypochord attracts precursors to the midline.50 In contrast, in zebrafish, while somite-derived VEGF-A secretion (induced by Shh in the notochord) is important for arterial specification of these angioblasts (reviewed in Ref.52), VEGF-A does not seem to be absolutely required for their migration from
the LPM to the midline.51 In addition, contact with endoderm causes the EC precursors to migrate as individual cells in zebrafish.51 Additional signals may assist in angioblast navigation to the midline (reviewed in Ref.49): semaphorins (e.g., semaphorin 3a1, a repulsive signal expressed in the somites53), CXCR4/SDF-1, and sucrose nonfermenting-related kinase (Snrk)-1.54 We recently identified the PDZ domain-containing scaffold protein synectin as a regulator of angioblast migration. Its absence in zebrafish resulted in angioblast stalling at the LPM or in between the LPM and the midline.55 In zebrafish, there are two waves of migrating angioblasts, one starting around 14 hours postfertilization (hpf), another around 16 hpf. While the first wave will give rise to the DA, the second wave will contribute to the PCV (see below). In avian—and most likely mouse but not zebrafish—embryos, in addition to the laterally migrating angioblasts, there is a second source of angioblasts, migrating ventrally from the posterior half of the somites to contribute to the DA in response to a yet unidentified signal56 (FIGURE 38.2). While the LPM angioblasts contribute to the floor of the primitive DA, this somitic angioblast population first invests the roof of the primitive DA and then spreads over the entire aorta to form the definitive DA. This coincides with the conversion of ECs in the floor of the aorta from hemogenic to nonhemogenic endothelium (see above)56 (FIGURE 38.2).
the LPM to the midline.51 In addition, contact with endoderm causes the EC precursors to migrate as individual cells in zebrafish.51 Additional signals may assist in angioblast navigation to the midline (reviewed in Ref.49): semaphorins (e.g., semaphorin 3a1, a repulsive signal expressed in the somites53), CXCR4/SDF-1, and sucrose nonfermenting-related kinase (Snrk)-1.54 We recently identified the PDZ domain-containing scaffold protein synectin as a regulator of angioblast migration. Its absence in zebrafish resulted in angioblast stalling at the LPM or in between the LPM and the midline.55 In zebrafish, there are two waves of migrating angioblasts, one starting around 14 hours postfertilization (hpf), another around 16 hpf. While the first wave will give rise to the DA, the second wave will contribute to the PCV (see below). In avian—and most likely mouse but not zebrafish—embryos, in addition to the laterally migrating angioblasts, there is a second source of angioblasts, migrating ventrally from the posterior half of the somites to contribute to the DA in response to a yet unidentified signal56 (FIGURE 38.2). While the LPM angioblasts contribute to the floor of the primitive DA, this somitic angioblast population first invests the roof of the primitive DA and then spreads over the entire aorta to form the definitive DA. This coincides with the conversion of ECs in the floor of the aorta from hemogenic to nonhemogenic endothelium (see above)56 (FIGURE 38.2).
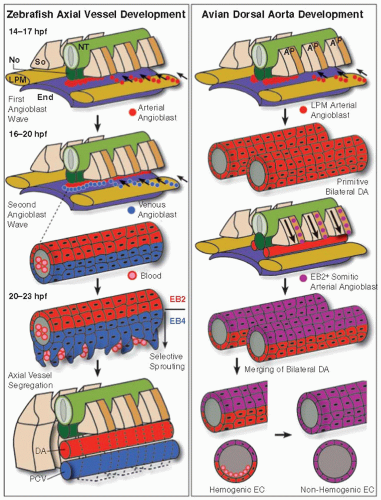 FIGURE 38.2 Vasculogenesis: the making of the main axial vessels. Left: Axial vessel formation in zebrafish. During early somitogenesis (14 to 17 hours postfertilization or hpf; somites indicated by “So”), the first wave of angioblasts (red circles) moves over the endoderm (End) from the lateral plate mesoderm (LPM) to the midline, just underneath the notochord (No). At some point during/after this journey, these angioblasts start to express ephrinB2a or “EB2” (under the influence of an arterial EC-inductive Shh-VEGF-Notch cascade) and give rise to a primitive vascular cord. Later (16 to 20 hpf), a second wave of LPM-derived angioblasts (blue circles, not expressing EB2) moves to the midline to join the cord. Subsequently (20 to 23 hpf), this common, mixed vascular cord gives rise to two axial vessels by selective sprouting, regulated by differential expression of EB2 and EphB4 (“EB4”) where EB4-expressing cells sprout ventrally to form the PCV, while the EB2-expressing cells stay put and form the dorsal aorta (DA). Red blood cells (with pink nucleus) invading the nascent PCV are associated with lumen expansion of the PCV. Right: Avian dorsal aorta development. In contrast to zebrafish, the DA is initially paired in avian embryos. The primitive paired DA forms by lateral movement and merging of LPM-derived angioblasts (red circles). Later, the roof of the DA is replaced with angioblasts (purple circles) coming from the posterior (“P”) part of the somites. These angioblasts express EB2 already at the onset of their journey. The paired chimeric DA then merges to form a single axial vessel. Around that time in the LMP-derived ventral part of the DA, “hemogenic” ECs give rise to hematopoietic cells (with pink nucleus), an activity that is terminated when the ventral part of the aorta is replaced by somite-derived ECs. NT, neural tube. (Adapted from Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature 2005;438(7070):937-945.) |
The next step following migration is the establishment of a tube/cord-like structure. In Xenopus, avian, and mouse embryos, endoderm-derived hedgehog signals (Shh in chicks; an unidentified hedgehog signal in mice) provide necessary cues for tube formation.57,58 Unlike its indirect effect (through stimulation of VEGF-A production) during arteriovenous specification and postnatal angiogenesis,59 Shh most likely directly interacts with EC precursors during tubulogenesis.58 Interestingly, however, regional differences seem to exist along the anterior-posterior axis of the DA of zebrafish. While in the posterior part of the DA, tube formation can occur in the absence of endoderm,51 a recent study showed that the formation of the bilateral DA (the anterior portion of the DA) is critically dependent on the endoderm-derived cxcl12b (a zebrafish orthologue for SDF-1).60 Analogous to what is known from epithelial tube formation,61 the establishment of vascular tubes involves at least three coordinated activities,62,63 that is, the increased cellular contact (mediated by junctional molecules, e.g., vascular tubulogenesis is severely affected in VE-cadherin knockout mice64), the establishment of apical-basal polarity (involving translocation of junctions to the lateral side, exocytosis to the apical surface of negatively charged antiadhesive proteins, such as CD34-sialomucins that form a cell-cell repulsive glycocalix, and rearrangement of the cytoskeleton), and interaction with the surrounding matrix (e.g., fibronectin51,65) through integrins (e.g., β1 integrins65,66). In the zebrafish, angioblasts from the first migration wave initially aggregate without the expression of adhesion molecules, then a fibronectin matrix is deposited between the cells, and zona occludens-1 (ZO-1) only marks cell-cell junctions 2 hours later at 17 hpf.51 Lumenization starts at 18 hpf according to a “hollow out” mode to form the future DA. Recently, it was shown in mice that lumenization of the DA is driven by VEGF-A that activates Rho-dependent kinases leading to cytoskeletal changes that open up a patent extracellular lumen between two apposing ECs.67 This extracellular mode of tube formation thus requires junctional rearrangement between ECs as opposed to another model based on fusion of intracellular vesicles within single ECs described for lumen formation during intersomitic vessel (ISV) formation in the zebrafish.68 The latter intracellular model has recently been complemented by another study showing that the lumens in the ISV of zebrafish are formed by rearrangement of ECs around an intercellular lumen.69 The mechanisms of vascular lumen formation thus seem context dependent.
The first signs of arteriovenous differentiation appear in the zebrafish around 17 hpf, upon expression of the arterial marker ephrinb2a in the cell aggregate that will form the DA.51 At this time, some cells in the ventral part of the cluster as well as those that constitute the second migratory wave do not express ephrinb2a and are therefore likely venous angioblasts. This is before the onset of circulation, meaning that this cell fate decision is independent of flow and hence genetically predetermined. When exactly arteriovenous fate decisions are made is not entirely clear. Similarly, in mice, polarized expression of ephrinB2 in the arterial cells and EphB4 in venous cells becomes apparent before the onset of blood circulation.70,71 While ephrinB2 expression studies suggest that LPM angioblasts only acquire arterial specific markers after migration, some molecules, such as synectin, seem to specifically affect arterial angioblast behavior during (or perhaps before) migration.55 Interestingly, the somitic angioblasts identified in birds and mice acquire ephrinB2 expression before/during their migration to the DA.56 By 24 hpf, both axial vessels (ephrinB2a+ DA and flt4+ PCV) are formed in the zebrafish trunk.51
The molecular cascades responsible for the induction of arteriovenous fate have been mostly determined in zebrafish and mouse embryos (including a Shh-VEGF-Notch cascade, TGFβ, Sox, Fox, adrenomedullin, Spreds, phosphatidylinositol-3, and COUP-TFII or chicken ovalbumin upstream promoter-transcription factor II). This process has been elaborately described in many comprehensive reviews.72,73,74,75 Due to space restrictions, here we only highlight the recent discovery that the Notch and Wnt/β-catenin pathways synergize to induce an arterial fate in vascular progenitors in vitro and in vivo.76,77
In mice, the size and segregation of the axial vessels seems to be critically determined by Notch and ephrinB2/EphB4 signaling, the former regulating the balance of arterial versus venous ECs and the latter “sorting” arterial and venous ECs into their respective vessels.78 A recent time-lapse analysis of main axial vessel formation in zebrafish confirmed such a sorting process whereby arterial and venous angioblasts first reside in a common vessel primordium at the midline. Subsequently, ephrinB2a/EphB4 reciprocal signaling regulates the selective ventral sprouting of venous ECs (which then form the PCV) and the restricted dorsal sprouting of arterial ECs (which then generate the ISVs)79 (FIGURE 38.2). Thus, according to this model, the PCV is not formed by vasculogenesis but rather by sprouting angiogenesis. Whether this is a characteristic of the zebrafish model or also applicable to other species remains to be determined.
Angiogenesis
Angiogenesis, or the generation of new endothelial-lined vessels from preexisting ones, expands and remodels the rudimentary vascular primordium resulting from vasculogenesis during development and most likely also is a (“the”) main mechanism by which new vessels arise after birth. Angiogenic mechanisms have been mostly unraveled by studying a few stereotyped forms of developmental angiogenesis, that is, the formation of the ISVs
in the zebrafish and the formation of the retinal vasculature in the mouse, the latter occurring after birth. Many of the events during development are recapitulated in adult angiogenesis. Furthermore, different modes of vascular expansion/remodeling can be distinguished: sprouting angiogenesis (or the formation of vascular branches mostly triggered by hypoxia or other stimuli), intussusception (or transluminal pillar formation; reviewed in Ref.80), and circumferential growth (which for instance occurs during collateral enlargement driven by shear forces, also known as “adaptive arteriogenesis.”81) Here, we review the process of angiogenic sprouting since our understanding of this process has significantly enhanced in the last decade.
in the zebrafish and the formation of the retinal vasculature in the mouse, the latter occurring after birth. Many of the events during development are recapitulated in adult angiogenesis. Furthermore, different modes of vascular expansion/remodeling can be distinguished: sprouting angiogenesis (or the formation of vascular branches mostly triggered by hypoxia or other stimuli), intussusception (or transluminal pillar formation; reviewed in Ref.80), and circumferential growth (which for instance occurs during collateral enlargement driven by shear forces, also known as “adaptive arteriogenesis.”81) Here, we review the process of angiogenic sprouting since our understanding of this process has significantly enhanced in the last decade.
Angiogenic sprouting is a dynamic and highly coordinated process during which ECs must perform distinct tasks and adopt specific functional identities81,82,83,84,85 (FIGURE 38.3): tip cell selection/sprout initiation, tip cell navigation/sprout guidance, stalk cell proliferation/sprout extension, tip-tip cell anastomosis/sprout fusion, pericyte recruitment/vessel stabilization, and “phalanx” cell conversion/return to quiescence. In addition, during angiogenic remodeling, initial arteriovenous fate decisions can be “reconsidered.” Again, VEGF-A takes a center stage position in this coordinated effort, with the help of another signaling pathway, the Notch pathway, which modulates VEGF signaling.86,87
Tip Cell Selection/Sprout Initiation
Expansion of the developing vasculature is largely driven by the need for oxygen in growing organs. Hypoxia, or the lack of oxygen, can trigger the local production of VEGF-A, which results in the creation of a VEGF-A gradient. The extracellular distribution of VEGF-A is additionally shaped by the existence of different splice isoforms with distinct matrix binding or diffusible properties, the bivalent VEGF165 isoform being the most crucial for correct angiogenic sprouting.88 In the retina, VEGF-A is secreted by astrocytes that move a little bit ahead of the expanding blood vascular network and are therefore indispensable for normal vascular expansion in the retina.89 Upon contact with higher VEGF-A concentrations, certain individual ECs expressing VEGFR2 nearest by the source of VEGF-A in the avascular area, called “tip cells,” flip their apical-basal polarity, send out long VEGFR2-expressing protrusions or “filopodia,” and break out of the frontline.
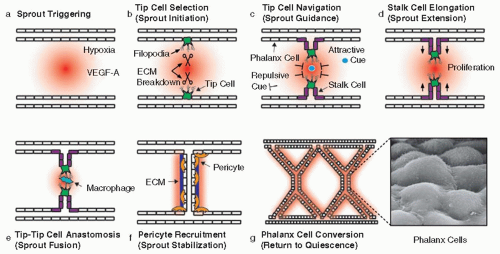 FIGURE 38.3 Angiogenesis: the different steps of the angiogenic ladder. Following a hypoxic trigger, a VEGF-A gradient arises (red; panel a). Next, cells encountering high VEGF-A concentrations change their polarity, break out from the vessel by breaking down the surrounding matrix, and develop filopodia to sense their immediate environment (panel b). These cells, “tip cells,” are guided on their way to another tip cell by attractive and repulsive cues (panel c). Cells following the tip cell (“stalk cells”) proliferate to elongate the sprout (panel d). Macrophages make both sprouting ends meet by finetuning tip cell guidance (panel e). After blood flow is restored, the new sprout is stabilized by deposition of extracellular matrix (ECM) and coverage with pericytes (panel f). The new vessel delivers oxygen to the tissue in need, thereby resolving hypoxia. As a result, the ECs revert to a quiescent phenotype, called “phalanx cells,” as they arrange themselves in a tight formation resembling the ancient Greek soldiers (panel g). (Adapted from Carmeliet P, De Smet F, Loges S, et al. Branching morphogenesis and antiangiogenesis candidates: tip cells lead the way. Nat Rev Clin Oncol 2009;6(6):315-326.) |
At the same time, VEGF signaling upregulates the Notch ligand delta-like 4 (Dll4). Dll4 subsequently activates Notch signaling in the neighboring cell, which results in downregulation of VEGFR2 and upregulation of soluble VEGFR1, a decoy receptor. As such, this neighboring cell becomes less sensitive to VEGF-A presented at the vascular front and becomes unable to upregulate Dll4. This lateral inhibition and self-reinforcing negative feedback loop thus induces a “stalk cell” phenotype in cells immediately adjacent to the tip cell and makes sure that tip cells retain their leading position. Detailed time-lapse analysis has however shown that tip/stalk cell identity is very dynamic and transient and that cells in a given sprout constantly compete, by differential expression of VEGFR1 and VEGFR2, for the tip position which involves repeated tip-stalk cell identity switches.90
In contrast to tip cells, stalk cells form fewer filopodia, but instead trail behind the leading tip cell to elongate the sprout (see below). Interestingly, these two distinct functional behaviors are reflected in a different expression profile: tip cells express high levels of VEGFR2, VEGFR3, Nrp1, UNC5b, Dll4, CXCR4, Ang2, ESM-1, SLP-76, and PDGF-BB, while stalk cells express more Jagged-1 (a Notch ligand), VEGFR1, and Robo4.87,91 Jagged-1 expression in stalk cells was recently shown to antagonize Dll4, thereby preventing Notch activation (and thus stalk cell induction) in neighboring cells. When modified by fringe glycosyl transferases, Notch1 is indeed activated more by Dll4 than by Jagged-1. Given that some Dll4 protein is detectable in stalk cells, Jagged-1 also helps to maintain differential Notch activity by antagonizing Dll4 that signals back to tip cells.92 In addition to Notch signaling, VEGF-A/VEGFR2 signaling in tip cells is further modulated by other mechanisms (reviewed in Ref.84) including cooperation with other VEGF receptors and a newly identified transcriptional repressor complex composed of translocation ets leukemia gene (Tel) and carboxy-terminal-binding protein (CtBP). Proximity ligation assays revealed that VEGFR2 can heterodimerize with VEGFR3 in tip cell filopodia upon VEGF-A or -C binding.93 The VEGFR2 coreceptor Nrp1, on the other hand, was shown to be involved in lateral filopodia extension, which is critical for tip cell turning and fusion (see below).94 Alternatively, Nrp1 alone may induce vessel branching by direct signaling in ECs through synectin.55 While VEGF coreceptors spatially modulate VEGF branching activity, the Tel-CtBP complex was shown to temporally restrict VEGF-A-stimulated Dll4 expression in sprouting ECs.95 Furthermore, this complex also regulated the expression of antagonistic sprouting cues (e.g., VE-cadherin and Sprouty4). In addition to their sensing behavior through filopodia, tip cells also need machinery to invade the surrounding basement membrane to be able to break out and advance. Matrix metalloproteinases (MMPs), such as MT1-MMP, may be one of the molecular tools to dissolve the matrix (FIGURE 38.3), at least during postnatal angiogenesis where the “mother vessels” from which the new sprouts are formed are embedded in a thicker layer of basement membrane.84
Tip Cell Navigation/Sprout Guidance
The tip cell filopodia function as sensors that constantly probe their environment. In this respect, tip cells resemble the growth cone of navigating axons.96 Therefore, certain guidance signals used by axons seem to have been co-opted by endothelial tip cells on their way to meet another tip cell82,97 (FIGURE 38.3). Tip cell movement needs to be correctly monitored by attractive and repulsive guidance posts. There are at least four ligand/receptor classes of guidance molecules with a documented role in vascular navigation: Slits/roundabout4, netrins/Unc5B, Semaphorin 3E/Plexin-D1, and ephrinB2/EphB4. Due to space restrictions, we only briefly illustrate some examples as prototypic examples. One roundabout (Robo) family member (Robo4) is expressed in growing vessels. Robo4 deficiency in mice leads to hypervascularization during oxygen-induced retinopathy, and its high expression in stalk cells may prevent these cells from being activated by VEGF-A.98 Mechanistically, Robo4 counteracts the leakagepromoting activity of VEGF-A by impairing VEGFR2-mediated activation of the kinase Src.98 The nature of the Robo4 ligand is debated, since Robo4 lacks Slit-binding domains, and Robo4 also binds to Unc5B, another guidance receptor, suggesting that Robo4/Unc5B maintains vessel integrity via Unc5B activation.99
Unc5B is highly expressed in tip cells, and binding of netrin-1 to Unc5B causes filopodia collapse in the retina while Unc5B knockdown in zebrafish induces ectopic ISV sprouting.100 Repulsion by netrin-1-Unc5B interaction requires the cytoplasmic domain of the receptor.101 However, netrin-1-deficient mice do not have a vascular phenotype, suggesting that netrin-1 signaling might involve other yet unidentified receptors or that other molecules may take over its role in mice.97 Alternatively, Unc5B could act as a dependence receptor that, in the absence of ligand, causes EC death.102
While secreted semaphorins usually bind to neuropilin receptors (functioning as a coreceptor for Plexins) and often inhibit vessel sprouting, Semaphorin 3E directly binds Plexin-D1 also causing vessel repulsion. Absence of either binding partner results in excessive branching in the trunk vessels of the mouse.103 Within the developing mouse retina, neuronderived Semaphorin 3E binding to Plexin-D1 activates RhoJ in ECs, thereby restricting and orienting VEGF-induced filopodia projection.104 In addition, this Semaphorin 3E/Plexin-D1 interaction fine-tunes the balance between tip and stalk cells by coordinating VEGF’s activity in a negative feedback.105 Similarly, the zebrafish mutant out of bounds maps to a mutation in the plexin-D1 gene which results in excessive and aberrant ISV branching into the somites; however, unlike in mice, the action of plexin-D1 in zebrafish is antagonized by Semaphorin 3E/plexin b2 signaling.106
Eph receptors and their ephrin ligands regulate cell contactdependent signaling through bidirectional signaling in cells expressing the receptor (forward signaling) or ligand (reverse signaling), in most cases generating repulsive signals.82,97 Notably, ephrinB2 was shown to regulate VEGFR2 endocytosis upon VEGF-A ligation during sprouting angiogenesis107,108 (reviewed in Ref.109). EphrinB2/EphB4 have also been shown to be involved in directional sprouting of ECs during the formation of the PCV in zebrafish (see above).79
Stalk Cell Proliferation/Sprout Extension
While tip cells lead the way, stalk cells follow the tip cell and are responsible for sprout elongation and lumen formation. Unlike tip cells that seldom proliferate, stalk cells do proliferate as a means to make the sprout longer (FIGURE 38.3). As mentioned, stalk cells acquire their phenotype by activation of Notch signaling. Since Notch signaling is generally known to inhibit EC proliferation,87 there must be another mechanism at play that overrules this inhibition. One of the downstream targets of Notch signaling in stalk cells is Nrarp which functions as a negative regulator of Notch through degradation of the Notch intracellular domain (NICD),110 thereby creating a permissive environment for proliferation. At the same time, Nrarp also activates Wnt signaling in stalk cells which stabilizes the stalk by inducing firm intercellular junctions.110 Instead of just passively trailing behind the tip cell, stalk cells may also actively push the sprout forward by secreting certain matrix components, for example Egfl7.111 As a result of Notch signaling, stalk cells have lower expression of VEGFR2 while they preferentially express VEGFR1.90 Notch activation also lowers endothelial expression of placental growth factor (PlGF), a VEGFR1-specific ligand.112 Gamma-secretase (which generates NICD by cleaving Notch) was shown to cleave VEGFR1, leading to release of its soluble form which can scavenge VEGF-A in the vicinity of the stalk cells, further dampening its effect.113 At the same time, the intracellular VEGFR1 fragment
may block VEGFR2 signaling. Collectively, these effects reduce and spatially restrict the overall response of ECs to VEGF-A and may fine-tune stalk cell morphogenesis. A recent paper revealed an additional fine-tuning mechanism of stalk cell behavior, whereby the response of an EC to Notch activation is negatively modulated through deacetylation—and hence destabilization— of NICD by SIRT1, an NAD+-dependent deacetylase.114
may block VEGFR2 signaling. Collectively, these effects reduce and spatially restrict the overall response of ECs to VEGF-A and may fine-tune stalk cell morphogenesis. A recent paper revealed an additional fine-tuning mechanism of stalk cell behavior, whereby the response of an EC to Notch activation is negatively modulated through deacetylation—and hence destabilization— of NICD by SIRT1, an NAD+-dependent deacetylase.114
Another task of cells in the stalk is the formation of a lumen, so blood flow can be initiated in the new sprout. Lumen formation in angiogenic sprouts may occur either before or after connection of the sprout to another tip cell83; it remains unknown how this occurs precisely. Possibly, lumen formation in anastomosing vessels mechanistically resembles that of vessels formed by coalescing angioblasts during vasculogenesis (see above).62
Tip-Tip Cell Anastomosis/Sprout Fusion
To make new connections, tip cells move toward each other. As they make contact, they need to convert their behavior of a migratory, navigating cell into a stalling connective cell. Recently, VE-cadherin, a junctional molecule, was shown to be expressed in filopodial protrusions of advancing tip cells, suggesting that interactions between cadherins of contacting tip cells may stabilize the connection.115 Formation of these junctions may then induce the phenotypic change from an actively navigating tip cell to a more quiescent cell. Failure to make such a stabilized contact, for example by repulsive forces between approaching tip cells, may on the other hand lead to sprout retraction, leaving behind an empty matrix sleeve. ECs may get assistance from helper “bridge” cells to acquire the precision needed to find another tip cell, as recently demonstrated in zebrafish where proangiogenic macrophages serve as guiding posts for tip cells and promote tip cell fusion116 (FIGURE 38.3). Recently, it was shown that VEGFR3 signaling in tip cells triggered by VEGF-C derived from macrophages positioned around sprout fusion sites results in fusion stabilization and phenotypic conversion of the fused tip cells to a stalk-like cell by reinforcing Notch signaling.117
Pericyte Recruitment/Vessel Stabilization
Maturation and stabilization of the nascent tube requires the deposition of extracellular matrix as well as the recruitment of pericytes (FIGURE 38.3). Pericytes, expressing PDGFRβ, are attracted to the endothelial tubes by PDGF-BB signals derived from the ECs. These EC-pericyte interactions additionally involve angiopoietin/Tie receptor signaling, S1P/sphingosine-1 phosphate receptor (S1PR) signaling, and Notch signaling (for comprehensive reviews118,119). Mural cell-secreted angiopoietin-1 activates its Tie2 receptor on ECs, thereby promoting pericyte adhesion to and junctional tightening between ECs. However, a recent study revealed that angiopoietin-1 seems less important for pericyte recruitment than originally thought and rather determines vessel diameter and number during vascular development.120 S1P1/S1PR coupling strengthens EC/pericyte contacts by relocating N-cadherin to the abluminal side of ECs.121 NOTCH3 mutations in CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) patients lead to vascular SMC degeneration and results in arteriopathy, which was recently phenocopied in mice.122
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


