3.1
Overview
The skeletal system consists of 206 bones of strikingly varying shapes, sizes, and functions. More than with any other organ, the specific shapes and sizes of these bones are crucial to their functions of providing levers for movement and protection of soft tissues. Despite the striking diversity of the sizes and shapes of individual bones, all bones form through one of two distinct processes: endochondral bone formation, used for the generation of most bones, and intramembranous bone formation, used to form the flat bones of the skull and parts of several other bones. In each of these processes, local paracrine signals and systemic hormonal signals trigger characteristic transcription programs and activation of kinase cascades that orchestrate the generation of the skeleton. In this chapter, we consider the strategies used to pattern the skeleton and then consider the processes of endochondral and intramembranous bone formation during development. Particular attention will be paid to description of the signaling systems, transcription factors, and cell types that coordinate the formation of the skeleton.
3.2
Patterning the skeleton
The skeleton is one of the most highly patterned structures in higher organisms. The 206 bones of the adult human vary greatly in size and shape; for instance, the femur may exceed 45 cm in length and its cylindrical shaft is remarkably different from the microscopic shape of ossicles of the inner ear. The anatomical aspect of bones also greatly differs between species; the skeleton of an elephant can be easily distinguished from that of a bat. However, the skeleton is patterned by mechanisms that are similar among all vertebrates. Our knowledge of these mechanisms largely comes from studies of chicken and mouse embryology.
Each tissue present in any organism originates from one of the three embryonic layers that are defined during gastrulation. The ectoderm is the outer most layer and is responsible for the formation of the tegument and associated structures, the epidermis, the nervous system, and some cranial bones. The inner most layer, the endoderm, forms the digestive track and associated glands, the lungs, the liver, and the pancreas. The mesoderm constitutes the intermediate layer and gives rise to the skeleton (with the exception of several cranial bones), the muscles, and some internal organs such as the kidneys. We commonly distinguish several types of mesoderm, which each form different derivatives, depending on the position of this tissue in the embryo along a radial axis ( Fig. 3.1 ). The paraxial mesoderm (PM) corresponds to the tissue immediately adjacent to the neural tube and to the notochord, tissues that form a central anteroposterior axis in the embryo. The PM will give rise to the axial skeleton (the vertebral column and associated ribs) and to some bones of the skull. The lateral plate mesoderm (LPM) corresponds to the mesoderm present furthest radially on each side of the neural tube; it will give rise to the appendicular skeleton (the limbs). Lastly, the intermediate mesoderm, located in between the paraxial and the LPM, does not give rise to any part of the skeleton but is necessary for the formation of internal organs such as the kidneys. While, with few exceptions, the embryonic origins of the all the bones are known, the molecular signals and tissue interactions required for patterning the skeleton still constitute an active field of investigation. We provide below, a summary of the complex sequence of events that generate the axial, craniofacial, and appendicular skeleton.
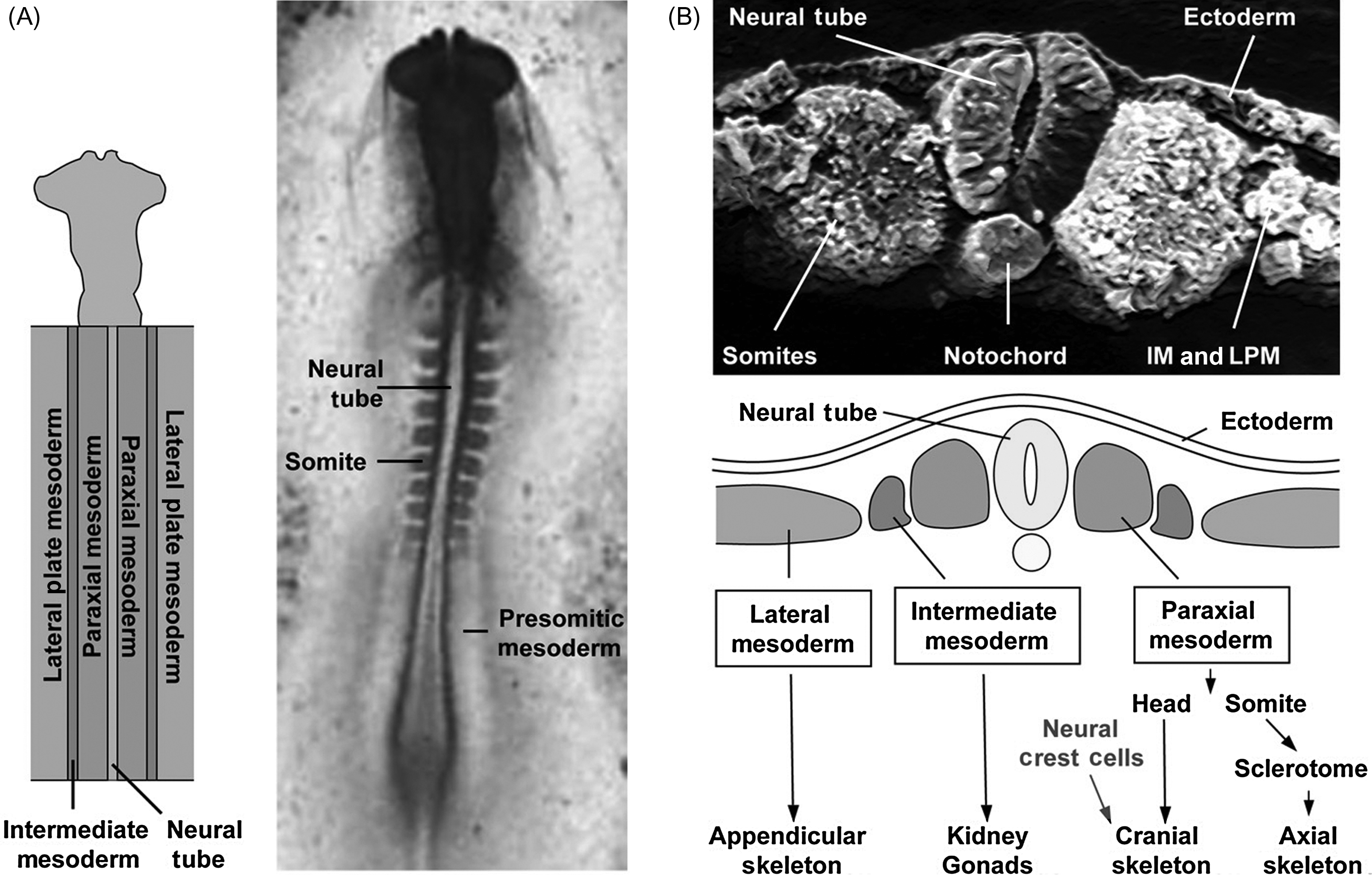
3.2.1
Axial skeleton development
The axial skeleton of vertebrates consists of the vertebrae and the intervertebral discs forming the vertebral column, and the ribs. The vertebral column is an essential element of support and motility of the vertebrate body. It provides attachment for many tendons/muscles in addition to the ribs and some organs, and it protects the spinal cord, which controls most bodily functions. It is also one of the most obviously segmented structures in animals.
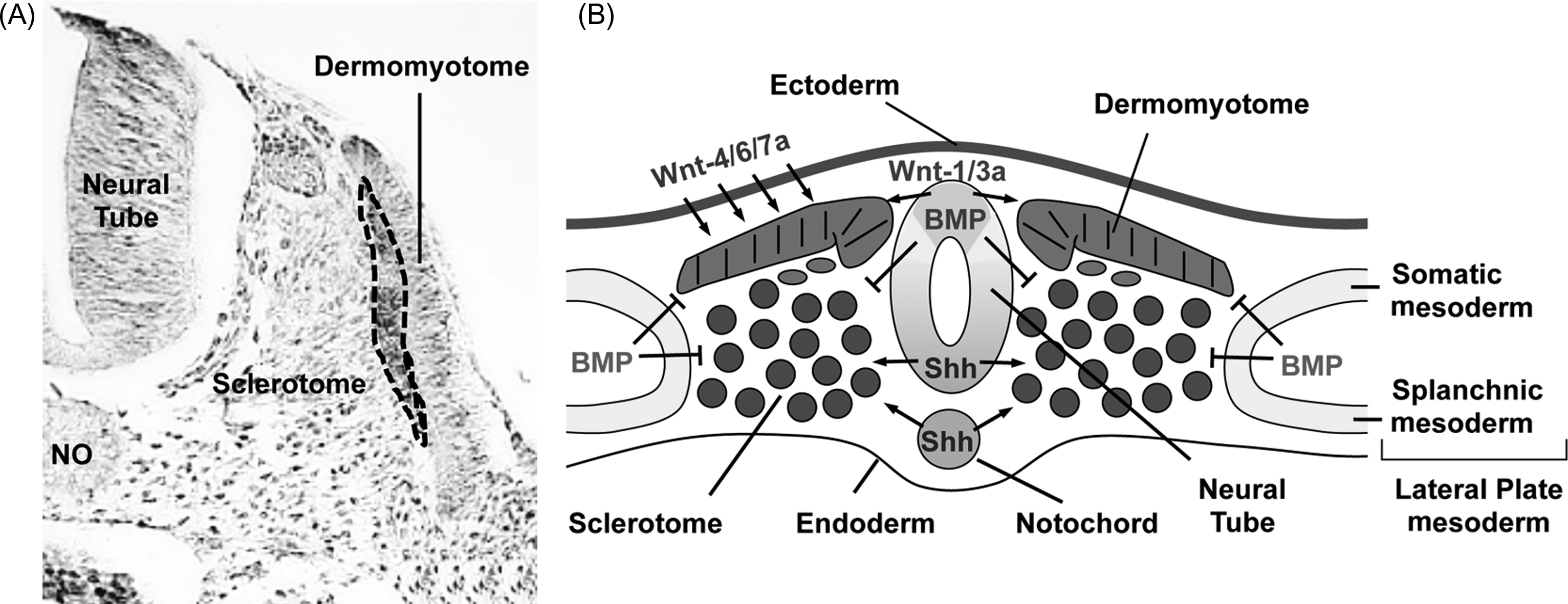
Formation of the anteroposterior axis of vertebrates occurs through a rostral-to-caudal progression of signals from the embryonic organizer known as the node and Hensen’s node in mammals and birds, respectively. These nodal signals result in the production of a variety of tissues including the PM that gives rise to the axial skeleton. The axial skeleton displays a metameric organization that consists of a series of equivalent units distributed along the anteroposterior axis, each comprising a vertebra and its associated muscles, peripheral nerves, and blood vessels. This segmental pattern is established during embryogenesis through the process of somitogenesis, by which the caudal mesenchymal portion of the PM, called the presomitic mesoderm (PSM), becomes segmented into epithelial somites, on each side of the neural tube ( Figs. 3.1 and 3.2 ). Fate-mapping studies have defined the progeny of different regions of the somite. Interspecies grafts of portions of the somite between chick and quail embryos allows the distinction of cells derived from the grafted tissue by histological staining (nucleolar quail-chick markers developed by Le Douarin ) or by immunohistochemical staining with species-specific antibodies. These experiments have established that each somite differentiates such that the ventral region becomes mesenchymal and forms the sclerotome, precursor of the vertebrae, and the medial portion of the ribs, while the dorsal region, termed the dermomyotome, remains epithelial and forms skeletal muscles of the back, body walls and limbs, the dermis of the back, a portion of the scapula, and perhaps the distal portion of the ribs, although this is controversial ( Fig. 3.3 ). Interestingly, the most distal portion of the ribs, called the sternal ribs, derives from the LPM, suggesting that ribs may derive from three different tissues (the sclerotome, dermomyotome, and LPM).
In addition to the dorso-ventral regionalization of the somite, the sclerotome is further subdivided into several compartments organized along rostrocaudal and medialateral axes, increasing the complexity of axial skeleton patterning. For instance, the medial part of the rostral sclerotome (media-rostral quadrant) forms the vertebral body, whereas the media-caudal quadrant leads to the intervertebral disk, and the latero-caudal quadrant leads to the neural arches, the pedicules of the vertebra, and the ribs . The rostrocaudal compartmentalization has strong implications for the formation of vertebrae. One vertebra is not produced from one sclerotome but rather from the fusion of the caudal half of the sclerotome of one somite with the rostral half of the following somite in a process called resegmentation . In contrast, each axial myotome that gives rise to the muscles of the back derives from a single somite. The consequence of this is that the muscles of the back are attached to two successive vertebrae, allowing the vertebral column to bend.
The last level of regionalization of the somitic mesoderm corresponds to a specification of this tissue according to the anteroposterior axis of the embryo. Thus the somitic mesoderm is patterned into cervical, thoracic, lumbar, sacral, and caudal regions. This regionalization is established early during development, probably before the formation of the PSM, and relies on a changing expression of a family of homeobox-containing transcription factors, the Hox genes, along the anteroposterior axis . Hox genes are sequentially activated in time and space, in a way that reflects their organization into clusters in the genome. This phenomenon is known as the principle of colinearity. The molecular control insuring the collinear expression of Hox genes in the body is still not completely understood . Interestingly, recent work shows that specific epigenetic regulations through nucleosome compaction, micro RNAs, and ribosomal proteins mediate both transcriptional and posttranscriptional controls of Hox expression adding a new layer of specificity and complexity in the control of the gene expression that patterns the vertebrate axial skeleton. The colinearity of expression of the Hox genes has been conserved during evolution, while the number of Hox genes, and, therefore, the complexity of the specific combinations of Hox genes expressed along the anteroposterior axis, has increased in parallel with the increased complexity of the organisms. Primitive fishes as well as more recent vertebrate animals evolved from a common invertebrate ancestor. It is estimated that the first spineless creatures that lived hundreds of million years ago had up to 13 Hox genes to direct their development. When the first mammals appeared much later, they had four sets of 13 Hox genes distributed on different chromosomes. Since then, some genes have been lost during evolution, and recent mammals, such as mice and men, have a total of 49 Hox genes distributed in four different clusters. Thus the regionalization of the somitic mesoderm, defined by the specific combination of Hox genes (the Hox code) expressed in specific regions along the anteroposterior axis can explain why some animals such as snakes have ribs all the way from the neck to the tail, whereas humans have only 12 pairs of ribs, attached to the thoracic vertebrae. The importance of this regionalization and the Hox code is demonstrated by a very simple experiment: when a piece of PSM dissected from the thoracic domain of PM (that will form the ribs) of a donor embryo is grafted into the cervical domain of the PM of a host embryo, this tissue later develops ectopic ribs in the cervical region .
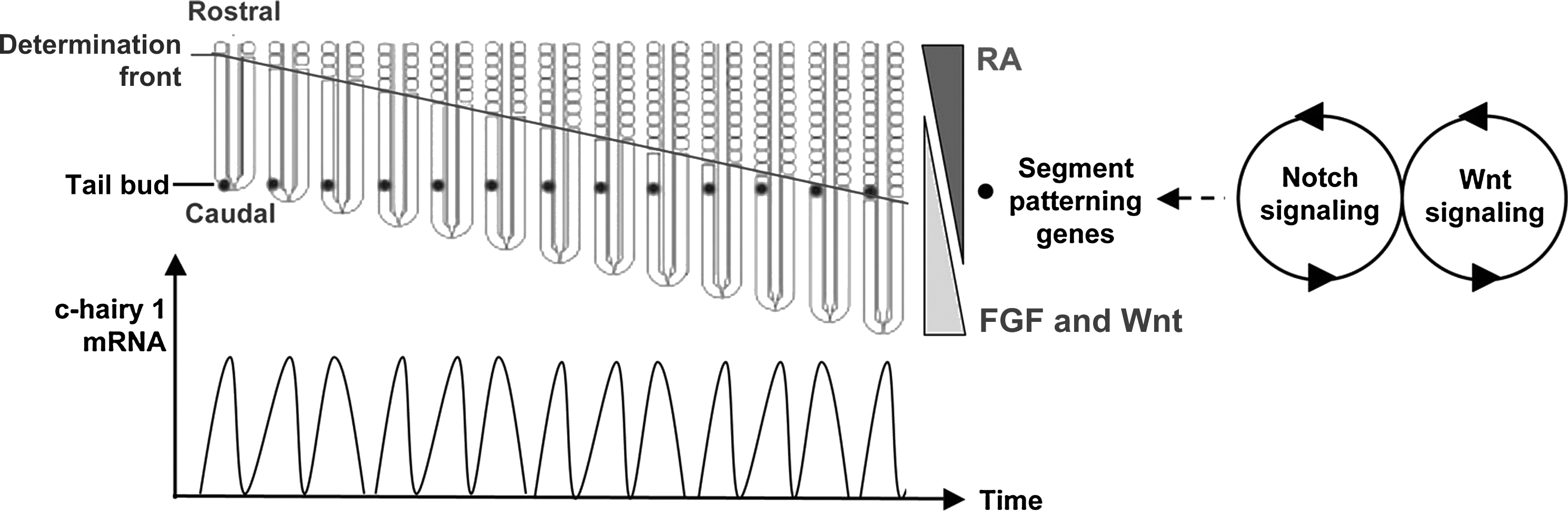
The process of somite formation is elaborately regulated ( Fig. 3.2 ). Somites form at a regular pace that is species-dependent: one somite is formed every 90 minutes in the chick and every 120 minutes in the mouse, in a rostral-to-caudal sequence. The segmentation of the PSM into somites relies on two distinct processes: the generation of a determination front that moves posteriorly during somitogenesis and the actions of an oscillating biological clock that determines the temporal periodicity of somite formation . This oscillator involves the periodic activation of the Notch and the Wnt signaling pathways . As in many biological clocks, such as the circadian rhythm, molecular oscillations are generated through negative transcriptional feedback loops. Recent work improved our understanding of the complex molecular mechanisms associated with the oscillations that activate the tissue segmentation machinery required for somitogenesis (reviewed in ). Notably, studies revealed that posttranscriptional regulations by miRNAs and proteasomal degradations are essential to control the clock , in addition to the transcriptional regulations characterized previously. While the segmentation clock controls the temporal periodicity of somite formation, the periodicity of the somite distribution in space is mediated by a moving front (or wavefront) of cell competence (called the determination front) that travels along the anteroposterior axis of the embryo. Two dynamic and antagonizing gradients of morphogens control the progression of this front along the anterior-posterior (AP) axis. Wnt and fibroblast growth factor (FGF) signals are generated rostrally and lead to gradients with decreased expression of these factors anteriorly; these gradients are opposed by a gradient of retinoid acid, which is synthesized maximally anteriorly. The position of the determination front has been proposed to be defined by a threshold level of FGF signaling, which has been suggested to be activated downstream of Wnt signaling. As the embryo extends posteriorly, the position of the determination front recedes along the anteroposterior axis. The classic “clock-and-wavefront” model argues that PSM cells become competent for segmentation when FGF signaling drops below a certain threshold, and then the cells adopt a boundary fate if they are juxtaposed to cells in a different phase of the segmentation cycle. This juxtaposition is realized when the wave of expression of cyclic genes sweeps the embryo from the tail to the head. Because of the oscillating nature of the cycling genes, this interface is transient, and a somite is, therefore, generated once per clock cycle. This classic view of the segmentation of the PSM implies that the clock and the wavefront are independent entities, and that only one phase of the clock is sufficient to determine the boundaries of the somites formed. Although the importance of the clock in somite formation has been recently challenged , data accumulated over the years overall strongly support the notion than intricate regulations governed by both clock genes and morphogen gradients are essential for PSM segmentation. Regardless of the classic “clock-and-wavefront” model or variations of this model that have been also described (see Ref. ), it appears clear that somitogenesis, and ultimately anteroposterior patterning of the embryo, requires a precise orchestration of multiple molecular signals responsible for the segmentation clock and the progression on the determination front. As a consequence, any alteration of these signals leads to severe segmentation defects that often cause axis truncation, and early embryonic lethality (reviewed in Ref. ).
A different group of signals specify the compartments of the somite (dermomyotome and sclerotome) ( Fig. 3.3 ). After segmentation, positive and negative signals released from the surrounding tissues induce the differentiation of the sclerotome . The notochord, and the ventral part (floor plate) of the neural tube, secrete the morphogen, sonic hedgehog (Shh), which leads to the formation of sclerotome, in addition to promoting growth and survival of somitic cells. Conversely, Wnt signals from the ectoderm inhibit chondrogenesis and promote dermomyotome formation dorsally. Lastly, bone morphogenetic protein (BMP) signals from the LPM and the roof plate of the neural tube antagonize Shh ventralizing activity. The BMP signals are antagonized by the presence ventrally of Noggin, a BMP inhibitor secreted by the notochord. The first molecular markers detectable at the time of somitic compartmentalization are the Shh target genes, Pax1, in the ventral part of the somite, whereas Pax3 expression is restricted to the prospective dermomyotome. Mice engineered to lack Shh fail to form vertebrae indicating that this factor is absolutely required for the formation of vertebrae in vivo. Several observations, however, suggest that both Shh and Pax1 may be required early in the formation of vertebral cartilage but that later steps of somitic chondrogenesis can occur in the absence of these signals. Sclerotomal cells that move dorsally to give rise to the neural arches stop expressing Pax1. Consistent with this observation, Pax1 knockout mice do not develop the ventral part of the vertebra but have normal neural arches .
BMP signals are required for the formation of the axial skeleton. Misexpression of noggin in the somite, for example, leads to truncation or loss of the vertebrae and the ribs . The requirement for BMP signals to allow sclerotome chondrogenesis stands in contrast to the observation that the same signals earlier inhibit the elaboration of most, if not all, somitic lineages, including sclerotome . Shh expression is the key to explaining these contrasting actions of BMPs. Sclerotomal cells that have been exposed to Shh signals become competent to initiate chondrogenesis upon subsequent exposure to BMP signals, whereas cells directly, exposed to BMP signals in the absence of Shh, no longer exhibit sclerotomal characteristics but express LPM markers instead . Shh induces the homeobox-containing transcription factor, Nkx3.2/Bapx1, and the high mobility group (HMG)-box containing transcription factor, Sox9; these transcription factors confer a chondrogenic response to BMP signals in the sclerotome .
The winged helix transcription factor Foxc2/MFH1 is critical for the formation of the whole vertebra, but other genes control the formation of specific parts of the vertebra. As already mentioned, the absence of Pax1 leads to only the absence of the ventral part of the vertebra. Lack of Nkx3.2/Bapx1 and Nkx3.1 leads to a total absence of the ventromedial part of the vertebra, as well as to hypoplasia of the neural arches . In contrast, Zic1 mutant mice exhibit defects primarily in the neural arches . These few examples indicate that complex and precise vertebral morphogenesis relies on an intricate pattern of expression of multiple genetic activities.
3.2.2
Craniofacial bone development
Craniofacial bones have both an ectodermal and mesodermal origin. Most of the cranial skeleton derives from neural crest (NC) cells , which have an ectodermal origin. Because the NC cells give rise to derivatives generally produced by the mesoderm, the tissue formed by these cells is called mesectoderm, which forms the ectomesenchyme (cranial NC-derived mesenchyme as opposed to the mesodermal mesenchyme). NC cells emerge from the dorsal midline of the neural tube and migrate extensively to form various derivatives, both in the trunk, and in the head of the embryo . In the head, NC cells migrate to colonize the pharyngeal arches (also called branchial arches) and other structures more rostral that are surrounded by a layer of ectoderm (including the neuroectoderm and the facial, or surface, ectoderm), in order to form the connective tissue associated with head muscles, tendon, bone, cartilage, and dermis. Modern techniques, such as vital dye (DiI) labeling, construction of chimeric embryos (quail-chick or quail-duck chimeras), and the discovery of NC-specific markers (Wnt1), have shown that the extensive NC-derived ectomesenchyme primarily occupies the ventral part of the vertebrate head, whereas mesenchyme derived from the mesoderm occupies the dorsal part of the head. Thus bones of the face and frontal bones derive from NC cells, whereas parts of the otic bones and occipital bones at the base of the back of the skull have a mesodermal origin. Evidence for the tissue origin of the parietal bones that form the skull vault is conflicting in the chick, but data based on tracking cells descended from those expressing Wnt1 indicate a mesodermal origin for parietal bones in the mouse . Most of the cranial bones are formed through an intramembranous process, with the exception that the occipital bones are formed through endochondral ossification.
Some of the principles underlying the patterning of the complex structure of the cranial bones have emerged . Depending on their precise origins, the NC cells themselves contribute to patterning in a cell-autonomous fashion. On the other hand, signals from the ectoderm and the endoderm to which the NC cells migrate also contribute to the patterning of the skull . The cell-autonomous properties of NC cells are illustrated by the behavior of NC cells that migrate from the neural tube in the trunk. These cells have an extremely low skeletogenetic capacity when compared to cranial NC cells . When trunk NC cells were transplanted to the head, these cells did not form cartilage or bone, unless the grafts contained only a small number of such cells . These observations raised the possibility that trunk NC cells may exert inhibitory effects, and that these effects are not strong enough when the number of grafted cells is too low to prevent the positive signals generated by the surrounding environment. Thus this experiment and others suggest that both the environment and a cell autonomous program are important in the patterning of the cranial bones . The surface ectoderm, the neuroectoderm, and the pharyngeal endoderm are important sources of craniofacial patterning information. In the chick embryo, when the frontonasal zone of the ectoderm is ectopically transplanted to a more dorsal or a more ventral position of the frontonasal prominence, this results in duplications of the upper or lower beak structures, respectively . In this experiment the transplanted ectoderm expresses two potent morphogens, fibroblast growth factor 8 (Fgf8), and Shh, which are able to reprogram the fate of NC cells at the transplant site. Similarly, the neuroectoderm has an important influence on patterning the NC cells into the craniofacial skeleton, since blocking of the Shh signal provided by this tissue leads to craniofacial syndromes such as holoprosencephaly . Lastly, the role of pharyngeal endoderm was demonstrated by the fact that the removal of this tissue results in the reduction or absence of some facial bones, whereas ectopic grafting of this tissue results in supernumerary lower jaws. At least some of the patterning information in the pharyngeal endoderm is mediated via Fgf signaling .
The role of Shh as a crucial craniofacial morphogen is particularly emphasized by the devastating effects in craniofacial development produced when Shh signals are inhibited in humans, birds, mice, and even fishes. Exposing avian embryos to cyclopamine, a potent inhibitor of the Hedgehog signaling pathway, can induce cyclopic defects that are characterized by a single central eye and no discernable nose. Humans with mutations in SHH or downstream effectors also may exhibit cyclopia. Further, retinoic acid signaling in the rostral head plays also an important function since blocking its signaling induces defects that are reminiscent of those induced by Shh inhibition . Another secreted molecule, bone morphogenetic protein 4 (BMP4) has been shown to play a role in the morphological variations of the beak observed in different species of birds . Ectopic overexpression of BMP or conversely, inhibition of BMP signaling induces an increase or a decrease of the size of the beak, respectively .
In addition to these secreted molecules, several transcription factors have been shown to play critical roles in the genetic control of the head morphogenesis, and thus craniofacial bones. In the craniofacial region, NC cells express the homeotic genes of the homeobox transcription factor (Hox) gene family, both before and after their migration to the arches . In the head, two different domains are defined by the presence or absence of hox gene expression in NC cells: NC cells located in the first branchial arch and anterior structures (anterior hindbrain, midbrain, and forebrain) do not express any Hox gene, whereas NC cells present in the second and more posterior branchial arches express Hoxa2 . Interestingly, quail-chick chimera experiments have shown that Hox-negative NC crest cells are the origin of the entire facial skeleton . Conversely, Hox-positive cranial NC cells are unable to generate any membranous bone. Induced loss of Hoxa2 expression in the second arch results in duplication of maxillary and mandibular structures, which normally arise from the first arch. By contrast, ectopic expression of Hox genes in rostral domains blocks the capacity of cephalic NC cells to differentiate into skeletal structures . It has been shown that NC cells expressing Hoxa2 are more constrained in their ability to respond to local cues from the epithelial environment compared to cells devoid of Hox gene expression . These results further illustrate the fact that both intrinsic genetic programs and environmental signals are important, and that they must work together to achieve proper cranial bone patterning. Other homeobox genes have been shown to pattern the cranial skeleton, as well. Mice lacking Distal-less homeobox gene 5 (Dlx5) and Dlx6 exhibit a duplication of the upper jaw and an absence of mandible .
Another important determinant of cranial patterning is a series of signals responsible for the generation of and the migration of the NC cells from the neural tube to specific locations. Several craniofacial malformations, called neurocristopathies, can be attributed to defects in the generation and migration of NC cells (reviewed in Ref. ). The tyrosine kinase receptor ErbB4, for example, is expressed in the neural ectoderm and has been shown to be required in cultured embryos for the proper migration of NC cells . Other factors involved in segmental and directional NC cell migration are reviewed by Minoux and Rijli . NC cells always migrate toward NC cell-free zones. Interestingly, it has been shown that this is achieved by cell–cell contact between NC cells, which triggers a localized activation of RhoA, leading to the inhibition of cell protrusions in this region . As NC cell migration is laterally limited by repulsive signals, contact inhibition results in NC cells only moving forward .
Although significant progress has been made in our understanding of the early processes involving complex gene and signaling regulatory networks that result in morphologically distinct craniofacial shapes and sizes, (with most studies focused on differential NC cell proliferation and differentiation to explain these variations), later mechanisms controlled by NC cell-derivatives that could contribute to craniofacial morphogenesis remain elusive. Interestingly, osteoclast-mediated bone resorption, which has been extensively studied in the adult appendicular skeleton, has recently been shown to play an important role in the control of jaw length . Quails that harbor a much shorter beak than ducks present significantly higher bone resorption activity during the development of the jaw. When a quail-derived NC mesenchyme destined to form the jaw skeleton is grafted onto a duck embryo, duck chimeras develop quail-like beaks. Inhibiting or activating bone resorption during development significantly lengthens or shortens the jaw. Hence, bone resorption constitutes a key patterning mechanism controlled by the NC mesenchyme at least in the jaw (see review in Ref. ). Further work is needed to determine whether bone resorption contributes more broadly to the patterning of all other craniofacial bones.
3.2.3
Appendicular/limb development
Despite the enormous variety of shapes, sizes, and functions of animal limbs, all vertebrate limbs are similarly organized into three domains distributed in a proximal-to-distal fashion. In the generic limb the stylopod is the most proximal domain of the limb (the closest to the body wall) and contains only one bone (humerus or femur). The zeugopod corresponds to the middle domain and generally contains two bones (ulna and radius, or tibia and fibula). The autopod represents the most distal part of the limb. It contains a variable number of bones (in the wrist, ankle, and digits) and thus corresponds to the most divergent part of the limb in the multiple existing vertebrates. Like the bones of the axial skeleton, but unlike most of the bones in the skull, bones in the limbs are formed through the process of endochondral bone formation.
The limbs originate from the LPM, which also give rise to parts of the scapula and pelvic bones. In response to complex molecular signals not completely elucidated, the limbs, two forelimbs and two hindlimbs, emerge from the body wall and are initially composed of undifferentiated mesenchymal cells covered by a layer of surface ectoderm. Mesenchymal cells actively proliferate and are exposed to proximal-distal (PD), AP, and dorsal–ventral (DV) signals responsible for patterning the limb. Later in limb development, a decrease in the mitotic activity of the cells destined to form bones correlates with the aggregation of these cells into mesenchymal condensations. This process precedes the differentiation of the mesenchymal cell population into chondrocytes, and into connective tissues such as tendon and muscle sheaths. The different muscles necessary for the limb movements are formed from cells that migrate from the lateral edge of the dermomyotome. Here we describe the main mechanisms involved in the induction and growth of the limb, its PD, AP, DV polarity, and we present briefly the processes of formation of mesenchymal condensations and joints, two phenomenon that ultimately dictate the number, and very likely the shape and size of bones.
3.2.3.1
Formation of the early limb
The limbs originate from the LPM as swellings in the body wall called limb buds. Limb buds are present at specific locations along the AP axis of the embryo. For a long time the model that prevailed described the limb bud as homogenous mesenchymal cells actively proliferating and covered by a layer of surface ectoderm. According to this model, the initial budding of the limbs resulted from a higher proliferation rate observed in the mesoderm that will form the future limb, compared to a lower proliferation rate in the flank mesoderm. This initial scheme was, of course, too simplistic and important works revealed a more complex molecular control of the limb initiation and outgrowth. Notably, a recent study invalidated the assumption that the limb emerges from a preexisting mesenchymal cell population. Using a linage tracing approach and epithelial and mesenchymal markers, the authors of this work demonstrated that mesenchymal limb progenitors result from an epithelial-to-mesenchymal transition (EMT) that takes place specifically in the presumptive limb fields . Cells of the coelomic epithelium are induced to transition to mesenchymal cells by Tbx5 and Fgf10 , two genes known to control limb initiation (see later). Hence, EMT rather than differential cell proliferation play an essential role in the initiation of the limb.
Among all tetrapods a conserved pattern of limb placement exists: the forelimbs develop at a position where cervical vertebrae transition to thoracic vertebrae, while hind limbs develop where lumbar vertebrae transition to sacral vertebrae. As for many other embryonic structures and organs, several lines of evidence suggest that limb buds are induced in the embryonic flank at precise positions that are determined by expression of a specific combination of Hox genes. For example, the specific combination of Hoxc6 , Hoxc8 , and Hoxb5 expression directs the formation of forelimbs. In different vertebrates, this combination is present at different levels in the trunk, and, in each case, this “Hox code” always correlates with the region of forelimb formation and is lost in limbless vertebrates. Interestingly, mice lacking the Hoxb5 gene have the shoulder girdle slightly shifted, an observation that confirms a role for Hox genes in determining the region that will form the limbs. Recent works demonstrated more directly the essential role played by Hox genes in limb positioning. Hox proteins were found to bind to and directly regulate the expression of Tbx5 , a master regulator of forelimb development (see later) . In another study, Hoxb4 was found to be expressed early in the presumptive forelimb region, whereas Hoxb7 and Hoxb9 were expressed posteriorly in the interlimb region of the embryo. Interestingly, when a dominant negative form of Hoxb5 was overexpressed during limb specification stages, Tbx5 expression was restricted posteriorly, whereas dominant negative versions of Hoxb7 and Hoxb9 lead to a posterior shift of Tbx5 expression, which resulted in a posterior shift of the forelimb . Comparative expression analyses between vertebrate species confirmed the prediction that the duration of Hoxb4 expression determine the diversity of limb position among tetrapods .
The molecular mechanisms responsible for limb bud induction are not completely understood. It is known that signals from several axial tissues medial to the LPM, including the intermediate mesoderm, are important for induction of the limb bud. However, signals from the ectoderm that forms the external layer of the bud also seem to play a role in the initiation of the bud. In both cases, FGFs, FGF8 and FGF10, are known to control this step, in cooperation with Wnt signaling. The current model is that FGF8 activates Wnt signals, which in turn can restrict the expression of FGF10 in the area of the LPM where the bud will form. Consistent with this model, targeted mutation of the FGF10 gene in mice results in the absence of the limbs, most likely as a consequence of the interruption of limb bud formation. Also, recessive mutations of the gene encoding R-spondin secreted ligand 2 (RSPO2) known to activate WNT signaling were found to cause in humans the tetra-amelia syndrome, which is characterized by a total absence of the four limbs .
Despite the fact that the two pairs of limb buds emerging from the embryonic flanks look very similar at the earliest stages of development, major morphological differences appear thereafter. Early experiments performed in the chick embryo demonstrated that the specification between either forelimb or hindlimb is established at the earliest stages of limb development, before the formation of the limb bud. When LPM cells that belong to the forelimb field are taken prior to limb budding and are transplanted into an ectopic location, the ectopic limb generated always develops as a forelimb. Later, several genes encoding transcription factors are exclusively expressed in forelimb or hindlimbs in multiple organisms. For instance, the T-box transcription factor, Tbx5, and the Hox transcription factors, Hoxc4 and Hoxc5, are specifically expressed in the presumptive forelimb area, as mentioned before, whereas the expression of Tbx4 and the Otx-related homeodomain factor, Pitx1, is restricted to the presumptive hindlimb. More importantly, loss of function and gain of function studies in mice and chick embryos, respectively, demonstrated the role of Tbx5 in determining the forelimb identity, and the role of Tbx4 together with Pitx1 in hindlimb identity. Beside these few genes, most of the key molecules in limb development are similarly expressed in both forelimbs and hindlimbs, and thus the sequence of events responsible for skeletal limb patterning is extremely similar in each limb.
3.2.3.2
Proximal-distal patterning of the limb
The apical ectodermal ridge (AER) is a specialized structure present at the distal tip of the limb bud and corresponds to a thickening of the ectoderm that runs along the AP axis of the limb bud, separating the dorsal side of the limb from the ventral side ( Fig. 3.4 ). Beneath the AER a zone of undifferentiated cells called the progress zone is responsible for most of the PD growth of the limb. The fundamental role of the AER in limb growth was demonstrated almost 60 years ago, by microsurgical removal of the AER from chick embryos. Interestingly, when the AER was removed early in development, only the proximal (stylopod) part of the limb was formed, whereas when it was removed later, only the autopod was absent. This experiment was particularly important, as it demonstrated that the region corresponding to the stylopod differentiates first, followed by that of the zeugopod, and then that of the autopod. The AER has been found to maintain proliferation and survival of the cells present in the progress zone by secreting growth factors of the FGF family. When beads coated with FGFs are implanted into chicken limb buds in which the AER has been ablated, limb development occurs relatively normally. Mice in which FGF4 and FGF8 have been conditionally inactivated in the AER do not form limbs.
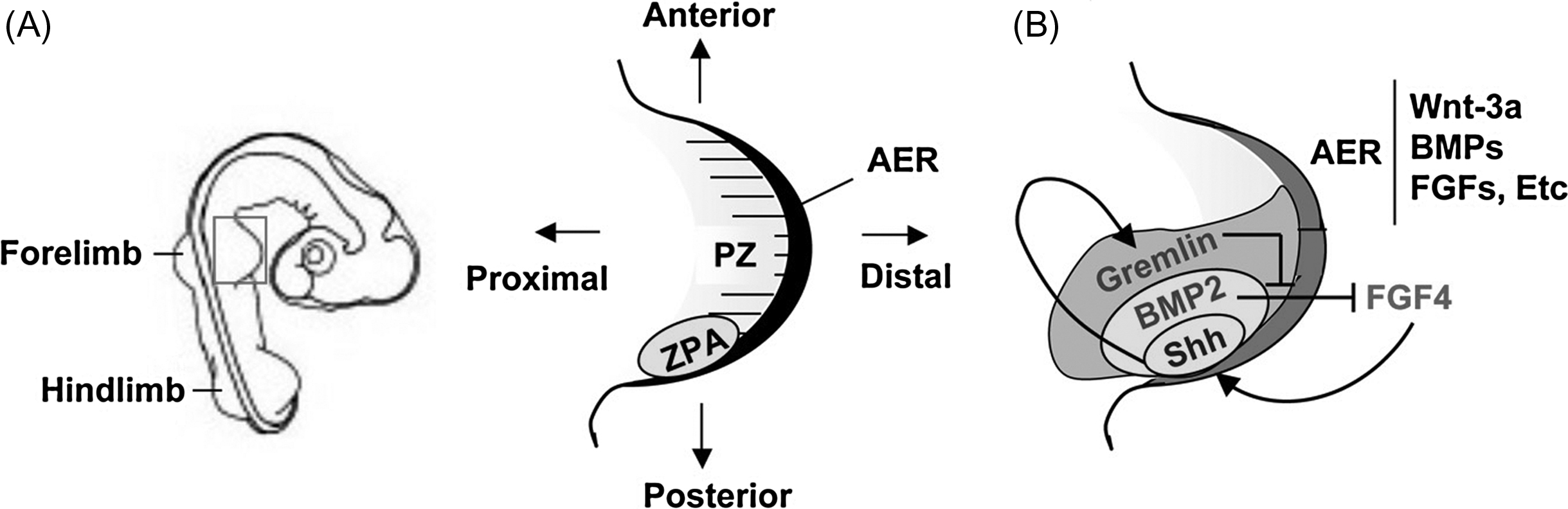
Until recently, one of the most actively debated issues regarding proximo-distal limb patterning was the question of how mesenchymal cells are specified to form the different structures of the limb skeleton (stylopod, zeugopod, and autopod). Two models had been proposed. The first model proposed was “the progress zone model” that postulates that the cells acquire positional information progressively, in a proximal-to-distal sequence. According to this model, cells present in the progress zone are subjected to signals from the AER for different period of times. As cells leave this zone progressively, their fate is determined by their time in the progress zone: the first cells leaving the progress zone have been exposed for a short period of time and become stylopod progenitors, cells leaving later are exposed longer to AER signals and become zeugopod progenitors, and cells leaving even later become autopod progenitors. Since its description, this first model has been challenged by fate-mapping studies that revealed that different groups of cells present at different depths within the progress zone contribute specifically to the formation of only one skeletal compartment. These observations led to a second model (the early specification model) that proposes that different groups of mesenchymal cells present in the early limb bud are already specified to form the stylopod, zeugopod and autopod; these cells proliferate to expand longitudinally and become determined to form particular structures in response to local signals. Current evidence favors the early specification model.
The understanding of how early specification of cell types occurs is incompletely understood. It has proven surprisingly difficult to differentiate between the autonomous and nonautonomous models for this specification experimentally. However, recent studies demonstrated that a dynamic balance between proximal retinoic acid signaling and distal FGF activity governs the PD pattern of major limb segments in the chick . These studies also showed that Wnt and FGF signals also combine to maintain the PD plasticity and the progenitor state of mesenchymal cells in the limb bud, a phenomenon reminiscent of what occurs during limb regeneration . Thus a balance of signals rather than a cell autonomous clock-like mechanism controls PD fates and establishes a differentiation front that is displaced distally (away from proximal retinoic acid influence) during progression of limb-bud outgrowth in the chick. However, one caveat to this instructive model comes from recent genetic studies suggesting that retinoic acid might be dispensable for PD limb patterning in mice . Directly comparable experiments in both species will be necessary to resolve this issue. Moreover, although most of the recent analyses suggest an instructive role for FGF in PD patterning, it will be also interesting to determine the precise role of FGF signaling in specifying each segment of the limb.
Recent studies (reviewed in Ref. ) have established a comprehensive model of digit patterning that proposes that iterative series of digits are established by what developmental biologists called a “Turing-like mechanism.” This mechanism is based on the idea that interactions between diffusible activator and inhibitor molecules that diffuse at differing rates from the same cell types lead to spontaneous emergence of periodic concentration profiles, generating specific self-organized patterns that prefigure the formation of digit and interdigit zones. Interestingly, the Wnt signaling agonist, Lrg6, marks stem cells of the nail that are present at the tips of the digits and is also responsible for digit tip regeneration in mice, in response to Wnt signals from the overlying epithelium . Hence, Wnt/β-catenin signaling also participates to the PD patterning of the limb.
3.2.3.3
The anteroposterior patterning of the limb
While the mechanisms determining PD patterning of the limb are thought to be distinct from those controlling AP limb morphogenesis (thumb to little finger), development along these distinct axes is coordinated. The zone of polarizing activity (ZPA) corresponds to a group of cells located in the posterior mesenchyme of the limb bud that acts as an organizer of the AP polarity of the limb ( Fig. 3.4 ). In chicken embryos, when the ZPA from one limb bud is grafted into the anterior margin of a host limb, a duplication of the digits is produced such that the ectopic digits form a mirror image of the normal digits present posteriorly. These observations were initially interpreted in terms of a morphogen gradient that diffuses across the limb bud to determine pattern in a concentration-dependent manner. The polarizing activity of the ZPA is mediated by the secreted factor, Shh. Ectopic expression of Shh in the anterior part of the limb bud mimics the effects of the ZPA grafts, suggesting that this factor plays an important function in polarizing the limb. Shh-null mice have limbs with more than simple AP axis abnormalities, however (reviewed in Ref. ). Shh-null limbs have preserved proximal structures (stylopod), but intermediate structures (zeugopod) are severely truncated and fused, while the autopod is almost completely absent. Thus Shh is not required to initiate limb development and is not involved in patterning the most proximal limb structures (stylopod). However, the dramatic abnormalities in PD patterning suggest that Shh acts to maintain the AER. In fact, Shh and FGFs form a regulatory loop between the ZPA and the AER ( Fig. 3.4 ). Indeed, removal of the AER leads to the loss of Shh expression, whereas the graft of cells expressing Shh in the limb bud induces ectopic FGF4 expression. Shh acts indirectly on the AER, through the induction of the expression of a BMP inhibitor, gremlin, made in the limb mesenchyme. Since BMPs suppress FGF4 expression in the AER, the net action of Shh is to stimulate the production of FGFs in the AER and maintain AER function. Thus Shh indirectly controls the PD development of the limb, in addition to its AP polarizing activity. Remarkably, deletion of both Shh and gli3, a transcription factor that mediates many of the actions of Shh, rescues the abnormalities of PD patterning in the Shh mutant alone and blocks the actions of Shh on AP patterning, as well. These data and others show that much of the action of Shh in the limb results from the suppression of expression of the repressor form of gli3 (Gli3R), which forms a gradient opposing that of Shh.
Several genes are known to control Shh expression in the ZPA. HoxD genes are expressed at specific times during limb morphogenesis in overlapping domains distributed along the AP axis to control Shh expression. In turn, Shh controls HoxD genes expression, thus forming a regulatory loop. The basic helix-loop-helix transcription factor, dHAND, also controls Shh expression in the limb bud. Mice deficient in dHAND expression die around E10.5 and present small limbs with no detectable expression of Shh. Lastly, retinoic acid has also been shown to control the AP polarization of the limb: inhibition of retinoic acid signaling prevents the establishment of the ZPA, the appearance of Shh expression, and the outgrowth of the limb bud, whereas grafts of beads coated with retinoic acid induce Shh and an ectopic ZPA in the limb bud. This effect of retinoic acid is thought to depend on the induction of Hox gene expression.
Several models for Shh-mediated AP limb axis patterning have been proposed (reviewed by Benazet and Zeller ). In the current model, progenitors, which will give rise to the digits (numbered 1–5 in mice, where 1 is the most anterior and 5 the most posterior), are exposed to varying amounts of Shh, and for varying times, depending on their position within the ZPA. In mice, while digit 1 is not exposed at all to Shh, digits 2 and 3 are exposed to low amounts of Shh for a short time, and digits 4 and 5 to a higher concentration of Shh for a longer time. Interestingly, though, genetic inactivation of Shh at defined developmental time points induces digit losses in a sequence that is not consistent with a morphogen gradient type of patterning mechanism . Instead, the sequence of digit loss follows the order by which digits normally condense. This study established that the specification of digit identities occurs within the first few hours of Shh signaling; subsequently, Shh is then continuously required to generate sufficient digit progenitor cells. Digit identities are then “fixed” (determined) substantially after specification and expansion of the digit progenitor domains. This is achieved by instructive BMP signaling coming from the interdigital mesenchyme, and targeting cells located at the distal tips of the forming digits. This region of mesenchyme located directly under the AER is called the phalanx-forming region (PFR) or digital crescent . Each PFR of a given digit is characterized by its unique BMP activity signature. Thus the degree of phospho-SMAD activity in PFRs determines digit identities. Moreover, FGF signaling from the AER also contributes to this late determination process by regulating the number of phalanges formed, which constitutes a hallmark of digit identities.
3.2.3.4
Dorso-ventral patterning of the limb
The dorso-ventral polarity of the limb is particularly evident when one considers epidermal-associated structures such as hair or feathers. In the case of the human hand the back of the hand is dorsal and the palm is ventral; muscles and tendons are found in an orderly pattern along this axis. Surgical manipulations involving the rotation of the ectoderm according to the DV axis have demonstrated that this tissue is responsible for determining DV polarity. Wnt7a is secreted by the dorsal ectoderm and controls the expression of the LIM-homeodomain factor, Lmx1, which is specifically expressed in the dorsal mesoderm beneath the ectoderm. Combined data from experiments involving ectopic expression in chicken embryos and targeted gene disruption in mice have demonstrated that Wnt7a and Lim1 are involved in the specification of the dorsal identity of the limb. Conversely, the expression of engrailed-1 (En1), which is restricted to the ventral ectoderm, is required for the specification of the ventral fate, since limbs of En1-null mice present structures ventrally that are similar to those normally observed only dorsally. Strikingly, En1-null limbs present ectopic digits ventrally, as a consequence of the formation of a second ectopic AER in the ventral limb, and demonstrate abnormalities of PD patterning as well. Thus the molecular signals that control DV patterning also indirectly affect the PD organization of the limb and, in that way, resemble the determinants of AP patterning.
3.2.3.5
Mesenchymal condensation and limb patterning
An important step in skeletogenesis is the condensation of mesenchymal cells, as this step is a prerequisite for chondrogenesis, which precedes bone formation in the limbs. Expression of the transcription factor, Sox9, is required for condensation, though its role in condensation is not understood. In vitro studies indicate that cell adhesion molecules such as N-cadherin and NCAM are important for the aggregation of mesenchymal cells. Several other molecules are expressed in mesenchymal condensations, including the growth and differentiation factor, GDF5 (a member of the BMP family), other secreted factors of the BMP, FGF, and Wnt families, the homeobox transcription factor Barx2, and Hox genes. FGFs may be important for stimulating SOX9 expression in condensations, since such stimulation is observed in mesenchymal cells cultured in vitro . The precise roles of FGFs and other factors, however, have not yet been demonstrated. Interestingly, a recent study demonstrated that, during tooth development, an inductive consequence of mesenchymal cell compaction induced by FGF8 can be mimicked by mechanical compaction and is mediated by the suppression of RhoA, a regulator of the cytoskeleton in response to mechanical forces . This observation raises the possibility that a mechanochemical control of mesenchymal condensation could also take place during limb, craniofacial, and axial skeletal development.
3.2.3.6
Joint formation and limb patterning
Joint formation occurs between two adjacent chondrogenic condensations, or within a single condensation, and constitutes another important mechanism for limb skeletal patterning since it determines the number and the size of some skeletal elements. The digits, for instance, initially constitute individual entities called digital rays (one per digit) that are then subdivided into phalanges through the process of joint formation. Typically, the prospective joint region is initially characterized by a group of cells that are denser and flatter than the chondrocytes present on both sides of this zone, called the interzone. Cells of the interzone then undergo cell death, which creates the joint space. Although not completely understood, the molecular and cellular mechanisms that lead to joint formation have been at least partly elucidated (see review Ref. ). As for the digit-interdigit patterning described earlier, the current model of the formation of the joints is based on a Turing-like mechanism, in which diffusible activator and inhibitor molecules create periodic self-organized areas that prefigure the formation of the synovial joints between phalanges. GDF5, a member of the BMP family, is strongly expressed in the interzone and is required for the formation of particular joints. Mice lacking GDF5 present several skeletal abnormalities including the loss of some specific joints in the autopod. GDF6 and GDF7 are also expressed in joints, but their expression is weaker and more restricted than that of GDF5. The loss of GDF6 function results in joint fusions, principally in the ankle and wrist, in sites distinct from those seen in GDF5 mutants. These results demonstrate that the GDF family plays a key role in establishing boundaries between skeletal elements during development, and they suggest that GDF members share the same task, with each of them determining a subset of joints. Deficiency in the BMP antagonist, Noggin, causes a failure of joint formation in the autopod, both in humans and mice. This failure may be due at least in part to an effect on GDF5 expression. In addition to BMP signaling, Wnt/β-catenin signaling plays a fundamental role in inducing joint formation. Ectopic expression of Wnt9a in chicken limb buds and mouse chondrocytes in vivo is able to induce GDF5 expression and ectopic joint formation, suggesting that this member of the Wnt family could play a physiological role in normal joint formation. Like GDF5, Wnt9a is an early marker of joint formation and has been shown to inhibit chondrogenesis and activate the joint-forming program. Recently, c-jun was shown to be required for the specification of the joints upstream of Wnt9a . Conversely, mice that lack β-catenin in chondrocytes present a lack of joint formation with the fusion of several bones. Interestingly, recent loss-of-function studies have shown that HoxD and several other factors can influence joint formation. This underlines the complexity of the molecular mechanisms that govern joint formation, and the pleiotropic roles that Hox genes exert in patterning the skeleton.
3.3
Endochondral bone development
As noted in Section 3.2 , the cranio-facial skeleton, the axial skeleton, and the skeleton of the limbs each begin as mesenchymal condensations. The sizes and shapes of these condensations, as well as the mechanisms controlling the variation in the sizes and shapes of the condensations, are regulated differently in each body region. Nevertheless, the next steps whereby mesenchymal condensations become bone follow one of only two fairly uniform processes: endochondral bone formation and intramembranous bone formation. We will consider endochondral bone formation first ( Fig. 3.5 ).
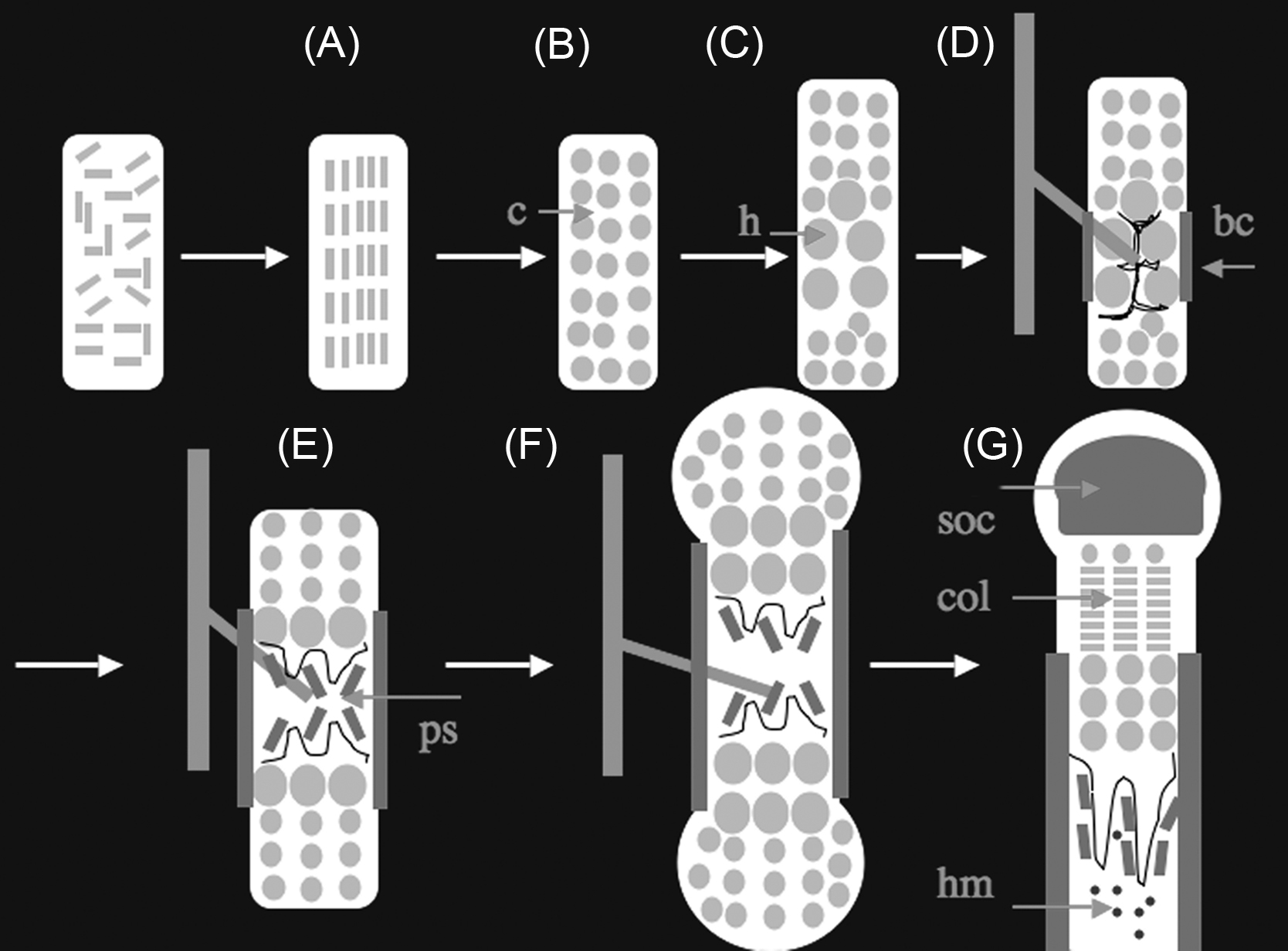
Mesenchymal cells in condensations differentiate into chondrocytes, round cells that secrete a matrix rich in collagen II and aggrecan. These chondrocytes proliferate, enlarging the bone anlage. In response to unknown signals, certain chondrocytes in the center of the anlage then stop proliferating, enlarge (hypertrophy), and change their genetic program to secrete a matrix rich in collagen X. These postmitotic hypertrophic chondrocytes direct the mineralization of the matrix surrounding them and signal to perichondrial cells to influence the differentiation of these cells into osteoblasts. The hypertrophic chondrocytes also signal to adjacent blood vessels to invade the bone anlage. Osteoclasts, cells of hematopoietic origin that can digest the extracellular matrix of bone or cartilage, also enter the bone anlage at this time. Hypertrophic chondrocytes then die an apoptotic death, leaving behind a mineralized matrix that serves as a scaffold for formation of a collagen I-rich matrix generated by osteoblasts. A fraction of these hypertrophic chondrocytes do not die, but, by a pathway not yet well characterized, can further differentiate into osteoblasts. Thus osteoblasts are descendants of both perichondrial precursors that can move into the marrow space with blood vessels and of hypertrophic chondrocytes. While this replacement of hypertrophic chondrocytes by osteoblasts occurs at the center of the bone anlage, chondrocytes further from the center of the bone continue to proliferate. The chondrocytes closest to the hypertrophic chondrocytes proliferate at a particularly high rate and flatten out, forming columns of flat proliferating chondrocytes that contribute to the asymmetric expansion of the cartilage mold. As bones enlarge further, secondary sites of ossification form by a mechanism that appears to repeat the process just described. In the center of an enlarging region of round proliferating chondrocytes, certain chondrocytes stop proliferating and become hypertrophic. This process is followed by vascular invasion and deposition of a bone matrix by osteoblasts that replace the hypertrophic chondrocytes that die through apoptosis. In the limbs the growth cartilage that remains between the primary and secondary ossification centers forms a disk of tissue called a growth plate. This growth plate continues to act as an engine for bone lengthening for characteristic periods postnatally. Depending on the bone and the species, growth plates can persist for life or disappear through poorly understood processes (called growth plate fusion). Here we will first consider the transcriptional mechanisms that regulate the differentiation and activities of chondrocytes, osteoblasts, and osteoclasts during development and then consider the signaling mechanisms responsible for the coordinated events of the endochondral sequence.
3.3.1
Chondrocytes
Chondrocytes are cells that produce and maintain a characteristic and abundant extracellular matrix. The cartilaginous matrix is comprised primarily of two components, the proteoglycans and the collagens . Proteoglycans are macromolecules containing a core protein with multiple attached polysaccharide chains. Because of their high content of charged polysaccharides, proteoglycans are highly hydrated. The polysaccharide chains in proteoglycans, called glycosaminoglycans (GAGs), are long repeating polymers of specific disaccharides. One of the most important extracellular proteoglycans is aggrecan, the predominant proteoglycan in cartilage. Aggrecan forms large aggregates; a single aggregate can be more than 4 mm long and has a volume larger than that of a bacterial cell. These aggregates give to cartilage its unique gel-like properties and its resistance to deformation. The GAGs covalently attached to aggrecan are keratan sulfate and chondroitin sulfate. Sulfation is thus an important translational modification in proteoglycan synthesis. The central component of the cartilage proteoglycan aggregate is a long molecule of hyaluronic acid. Hyaluronic acid is a large polysaccharide that forms a highly hydrated gel. Hyaluronic acid is bound to aggrecan in a noncovalent fashion and is the only extracellular oligosaccharide that is not covalently linked to a protein. The binding of hyaluronic acid to aggrecan is facilitated by link proteins that bind to the aggrecan core protein and to hyaluronic acid.
There are two different types of collagens in the growth plate matrix:
- 1.
Fibrillar collagens: collagen type II and collagen type XI. Collagen type II is found also in the vitreous of the eye. In cartilage, collagen type II is produced by proliferating chondrocytes and by the upper hypertrophic chondrocytes. Collagen type II is the most abundant collagen of the cartilaginous matrix. Collagen type IX is also found in the vitreous and binds GAGs.
- 2.
Sheet–forming collagen: collagen type X. Collagen type X is exclusively expressed by hypertrophic chondrocytes.
Several human genetic disorders associated with defects in either aggrecan or the transport of sulfate into the cells have been identified . Likewise, numerous chondrodysplasias are caused by mutations in each of these collagen genes .
Chondrocytes originate from condensed mesenchymal cells. Many lines of evidence have shown that Sox proteins are the master transcription factors for chondrogenesis . Sox9, as well as L-Sox5, and Sox6, are members of the Sox family of transcription factors that are characterized by the presence of the HMG-box DNA binding domain. Sox9 is required during sequential steps of the chondrocyte differentiation pathway; it is critical for commitment of mesenchymal cells toward the chondrocyte lineage, it upregulates expression of critical cartilaginous matrix genes such as those encoding collagen type II, collagen type IX, collagen type XI, collagen type X and aggrecan, and it prevents hypertrophic chondrocytes from transdifferentiating into osteoblasts, the cells forming bone . Several genetic approaches in the mouse (gain as well as loss of function) have demonstrated that Sox9 maintains chondrocytes in the proliferative state, blocks hypertrophic cells from choosing an osteoblast fate, and participates in the stimulation of collagen X gene expression in prehypertrophic chondrocytes . In humans, heterozygous missense mutations resulting in haploinsufficiency for expression of the Sox9 gene cause campomelic dysplasia, a rare disorder of skeletal development that results in deformities of most of the bones of the body . Most affected babies die from respiratory failure due to poorly formed tracheal and rib cartilage.
Two other members of the Sox family, L-Sox5 and Sox6, are required for chondrogenesis. Although individual L-Sox5 or Sox6 knock-out mice are born with minor cartilage defects, double knock-out animals develop a severe, generalized chondrodysplasia characterized by a virtual absence of mature cartilage, secondary to a defect of cell proliferation and impairment of cartilage matrix production . Similarly to Sox9, L-Sox5 and Sox 6 also control sequential steps of growth plate chondrocyte differentiation . The expression of L-Sox5 and Sox6 requires Sox9, and Sox9 may directly cooperate with these transcription factors in controlling the expression of cartilage matrix genes. Notably, both L-Sox5 and Sox6 lack the transactivation or transrepression domains and may thus act mainly to facilitate organization of transcription complexes.
Major regulators of chondrocyte hypertrophy include transcription factors of the Mef2 and Runx families . In particular, Mef2c is required for Runx2 gene expression in prehypertrophic chondrocytes; these transcription factors together are required for chondrocyte hypertrophy and collagen X expression. Histone deacetylase 4 (HDAC4) shuttles between the cytoplasm and nucleus in a regulated fashion, and, when in the nucleus, blocks the actions of Mef2c and Runx2. Runx2 belongs to the Runt transcription factor family and was initially characterized as a molecule essential for osteoblast differentiation . Inactivating mutations of human Runx2 cause cleidocranial dysplasia (CCD) in the heterozygous state. More detailed investigations have revealed that Runx2 is also expressed in chondrocytes as they initiate hypertrophy, and loss of this factor in genetically engineered mice severely delays chondrocyte maturation in a number of developing bones . Independent of its role in chondrocyte hypertrophy, Runx2 plays a critical role in vascular invasion of cartilage, as there is almost no vascular invasion in most skeletal elements of Runx2-deficient mice . Both the lack of expression of the angiogenic factor, VEGF (vascular endothelial growth factor), which is normally expressed in hypertrophic chondrocytes, and the observation that Runx2 binds to and activates the VEGF promoter in vitro suggest that VEGF mediates the Runx2-dependent regulation of blood vessel invasion .
The mammalian fetal growth plate is a virtually avascular tissue, but it requires an angiogenic switch in order to be replaced by bone. It is also a highly hypoxic tissue. The transcription factor, hypoxia-inducible factor 1 (HIF-1), is the major mediator of response to hypoxia in mammalian tissues and belongs to the PAS subfamily of bHLH transcription factors . One target of HIF-1α is VEGF-A.
Both HIF-1α and VEGF pathways are crucial for chondrocyte survival during development. In agreement with its angiogenic function, the conditional knock-out of VEGF in chondrocytes results in delayed blood vessel invasion . More surprisingly, the lack of VEGF generates massive cell death in the epiphyseal regions of the bones, in both the resting and proliferating zone of the growth plate . A similar phenotype is also observed in growth plates lacking HIF-1α . Taken together, these data indicate that VEGF and HIF-1α are components of a pathway that supports chondrocyte survival during endochondral bone formation.
Numerous lines of evidence suggest that chondrocytes and osteoblasts originate from a common osteochondroprogenitor and that activation of the transcription factor beta-catenin induces osteoblastic and suppresses chondrocytic differentiation in early osteochondroprogenitors . β-catenin is the key downstream signaling molecule of the Wnt canonical signaling pathway. Conditional deletion of β-catenin in limb and head mesenchyme during early embryonic development results in arrest of osteoblastic differentiation and lack of mature osteoblasts in membranous bones. Furthermore, in the absence of β-catenin, osteochondroprogenitors differentiate into chondrocytes instead of osteoblasts. Sox9 can bind β-catenin and block its actions. Thus the opposing actions of SOX9 and β-catenin help determine the commitment of osteochondroprogenitors and the pace of differentiation of chondrocytes.
Notably, chondrocytes with mesenchymal stem-like properties are present in the postnatal growth plate. This finding could pave the way to the identification of strategies that could be used to appropriately modulate that novel pool of stem cells and, consequently, growth plate development .
3.3.2
Osteoblasts
During intramembranous bone formation, osteoblasts derive from condensed mesenchymal cells . The origin of osteoblasts in endochondral bone formation is more complex. Osteoblasts that form the cortical bone (called the bone collar in embryonic life) differentiate from the mesenchymal cells of the perichondrium; a fraction of these cells, while still early in the osteoblast lineage, migrate along with blood vessels into the developing marrow space and form the primary spongiosa, the precursor of what will become trabecular bone . Periosteal skeletal stem cells also remain in the periosteum in adulthood . In addition, particularly during fetal life and the rapid phase of growth postnatally, hypertrophic chondrocytes have the potential to contribute substantially to the osteoblasts in the metaphyseal region of growing bones in mice . What relationship these osteoblast precursors from the perichondrium and growth plate have to skeletal progenitors in the bone marrow of adult bones is an unanswered question. Adult bone marrow contains cells capable of forming colonies in vitro, and these cells can also become osteoblasts, adipocytes, and chondrocytes (after fracture) and also support hematopoietic stem cells in vivo . These so-called CAR cells (CXCL12-abundant reticular cells) are a major source of osteoblasts in adulthood. Postnatal bones of mice also contain stem cells marked by the expression of Sox9 that can become CAR cells, osteoblasts, adipocytes, and chondrocytes (after fracture) . Independently of their origin, mesenchymal cells require three critical transcription factors in order to become osteoblasts during embryonic development, β-catenin (as already noted), Runx2, and Osterix . Committed osteoprogenitors cells then proliferate, differentiate into postmitotic osteoblasts that synthesize and mineralize bone matrix, and finally become terminally differentiated osteocytes or quiescent bone lining cells. Osteoblast differentiation is characterized by a loss of proliferative capacity and by a sequential increase in the expression of characteristic proteins, such as alkaline phosphatase, bone sialoprotein, collagen type I, PTH/PTHrP receptor, osteopontin, and finally osteocalcin and matrix metalloproteinase 13 . The mature osteoblast is found adjacent to the bone surface and has morphological and ultrastructural properties that are typical of cells engaged in secretion of a connective tissue matrix. Osteoblasts lay down bone matrix (osteoid) that is composed predominantly (90%) of collagen type I, along with noncollagenous proteins such as osteocalcin, osteopontin, osteonectin, and growth factors. After synthesizing and secreting the bone matrix and directing its mineralization, osteoblasts die or undergo two alternative fates: on quiescent bone surfaces, single layers of flattened, inactive osteoblasts are called bone lining cells. Alternatively, osteoblasts undergo a dramatic change in morphology and become buried in bone matrix as osteocytes . Osteocytes develop by forming numerous dendrite-like cytoplasmic processes that connect with adjacent cells to ensure their viability within the mineralized osteoid and to allow signaling. The cell bodies of osteocytes are found in lacunae, and the numerous processes lie in canaliculi. Osteocytes no longer synthesize collagen and appear to function as mechanosensors; they can reside in healthy bone for a long period of time, but in aging bone, empty lacunae are observed, suggesting that osteocytes undergo apoptosis . Osteocytes secrete modulators of osteoblast activity such as RANK ligand, osteoprotegerin (OPG), and sclerostin , suggesting the possibility that osteocytes directly or indirectly regulate the activity of osteoclasts and osteoblasts.
The transcription factor Runx2 is absolutely required during embryonic development for differentiation of mesenchymal cells into osteoblasts throughout the skeleton, both during endochondral and intramembranous ossification. Runx2-deficient mice have a cartilaginous skeleton without any osteoblasts, as their differentiation is arrested as early as E12.5 . In mice lacking only one allele of the Runx2 an abnormality in osteoblast differentiation is limited to bones formed through intramembranous ossification . A similar phenotype has been reported in humans with heterozygous loss-of-function mutations of Runx2 and is called CCD . CCD is an autosomal dominant condition characterized by hypoplasia of the clavicles, patent fontanels, supernumerary teeth, short stature, and changes in skeletal patterning and growth. Runx2 activity is controlled by various extracellular signaling pathways; the activity and stability of Runx family members are modified by phosphorylation, acetylation, and ubiquitination . Mitogen-activated protein kinase (MAPK) and PKA can phosphorylate and thereby activate Runx2 . Schnurri-3, a mammalian homolog of the drosophila zinc finger adapter protein, Shnurri, promotes Runx2 degradation , and the lack of Schnurri-3 leads to a dramatic increase in postnatal bone formation, at least in part by increasing the levels of Runx2 protein . Runx2 target genes include genes expressed by mature osteoblasts, such as osteocalcin, bone sialoprotein, osteopontin, and collagen type I . Runx2 expression is directly or indirectly regulated by the homeobox transcription factor, Msx2. Msx2 inactivation in mice causes a marked delay of ossification in the bones of the skull . (See discussion of intramembranous bone formation later.) This phenotype is concomitant with downregulation of Runx2 expression. Notably, one human syndrome characterized by increased bone formation at cranial sutures, Boston-type craniosynostosis, is caused by activating mutation in Msx2 . Paradoxically, in vitro, Msx2 can suppress Runx2 promoter activation ; thus the actions of Msx2 in regulating Runx2 are likely to be multiple and context-dependent. In an in vitro model of differentiation of early mesenchymal cells, for example, forced Msx2 expression stimulates these cells along the osteoblastic pathway and away from the adipocytic pathway, for example, Ref. . Thus Msx2 may act early in the osteoblast pathway to increase the number of osteoblast-specific cells.
Runx2 action is required for the expression of osterix, a zinc-finger protein related to Sp1 that is expressed in late chondrocytes and osteoblast progenitors . Like Runx2, osterix is required for the differentiation of osteoblasts. Osterix binds the transcription factor, Dlx5, and then binds to Dlx5-binding sites in the genome, activating a series of osteoblastic genes .
Another major regulator of osteoblast differentiation is the transcription factor, ATF4. ATF4-deficiency results in delayed bone formation during embryonic development . Though Col Ia1 gene transcription does not depend on ATF4, the synthesis of collagen I protein is dramatically lowered in ATF4 knockout mice, because amino acid availability is limiting. A major action of ATF4 is to regulate transporters that move amino acids into osteoblasts. Strikingly, high levels of amino acids in tissue culture medium correct the defect in collagen synthesis in ATF4 (−/−) osteoblasts, and a high protein diet prevents the osteopenia in ATF4 (−/−) mice . ATF4 also regulates the expression of genes expressed late during osteoblast differentiation, such as that encoding osteocalcin.
3.3.3
Osteoclasts
Osteoclasts are multinucleated cells that uniquely degrade mineralized matrix . Cells of the osteoblast lineage direct the differentiation and activation of osteoclasts by expressing the key regulators of these processes, M-CSF and RANK ligand (RANKL). Insights into the signaling pathways regulating osteoclast development have come from studying genetically altered mice with osteopetrosis, a condition characterized by the failure of bone resorption due to defective osteoclastogenesis or defective action of mature osteoclasts. Studies of osteopetrotic mice have made clear that osteoclasts are essential for resorption of the matrix left behind by dying chondrocytes during endochondral bone development. A characteristic finding in osteopetrotic bones is persistence of cartilage matrix remnants in what should be the marrow space. The spontaneous mutant op/op mouse displays an osteopetrotic phenotype with impaired osteoclast differentiation. In the complete absence of osteoclasts, no marrow space for hematopoiesis is formed, though vascular invasion and production of bone adjacent to the growth plate (primary spongiosa) still occurs . The mutation occurs in the gene encoding the cytokine, macrophage colony-stimulating factor (M-CSF), and calvarial osteoblasts from these mice cannot support osteoclast development when cocultured with spleen cells . The addition of recombinant M-CSF can restore bone resorption in vivo , and supplementation of cocultures with M-CSF results in the formation of osteoclasts in response to 1,25(OH)2-vitamin D .
RANKL is a type II transmembrane protein of the TNF family . It is expressed at highest levels in the bone and bone marrow but is also found in lymphoid tissues. Recent reports suggest that osteocytes may be a physiologically important source of RANKL . A soluble fragment of RANKL missing its transmembrane domain, along with M-CSF, is sufficient to induce osteoclast formation in the absence of supporting osteoblasts or stromal cells, suggesting that these proteins are the crucial osteoclastogenic factors produced by cells of the osteoblast lineage . RANKL expression is strongly stimulated by known activators of osteoclasts, including parathyroid hormone (PTH), IL6, IL11, and 1,25(OH) 2 vitaminD 3 . RANKL-null mice develop osteopetrosis with occlusion of the marrow space; they lack differentiated osteoclasts but do have precursors that can differentiate normally when cocultured with wild-type cells of the osteoblast lineage. In addition to defects in tooth eruption and mammary gland development, RANKL-deficient mice lack lymph nodes and demonstrate impaired differentiation of B and T lymphocytes .
The sole receptor for RANKL, found on osteoclasts and their precursors, is RANK, a member of the TNF receptor family . Polyclonal antibodies against the RANK extracellular domain can induce osteoclast formation in spleen cocultures when M-CSF is present . Furthermore, an anti-RANKL antibody lacking the Fc domain markedly inhibits RANKL-mediated osteoclastogenesis . As expected, mice with a targeted deletion of RANK have osteopetrosis. As with RANKL-deficient mice, mice lacking RANK have defective B and T cell maturation and lack peripheral lymph nodes, although thymic development proceeds normally .
OPG is a key modulator of activation of RANK by RANKL. OPG is a soluble member of the TNF receptor family . Production of OPG is strongly upregulated by estrogen, TNFα, GH, and TGFβ, while it is suppressed by PTH and glucocorticoids. In coculture experiments, OPG potently inhibits osteoclast induction by vitamin D, PTH, PGE2, or IL-11 . Hepatic overexpression of OPG in transgenic mice results in osteopetrosis with impaired thymocyte development , while administration of OPG to rodents results in dramatic increases in bone density with decreased osteoclast number and reduced serum calcium levels . Conversely, mice with a targeted deletion of OPG have severe osteoporosis due to increased bone resorption and increased numbers of osteoclasts . This finding demonstrates the important role of OPG in normal bone remodeling. Interestingly, the OPG knockout mice also have significant calcifications of the aorta and renal arteries .
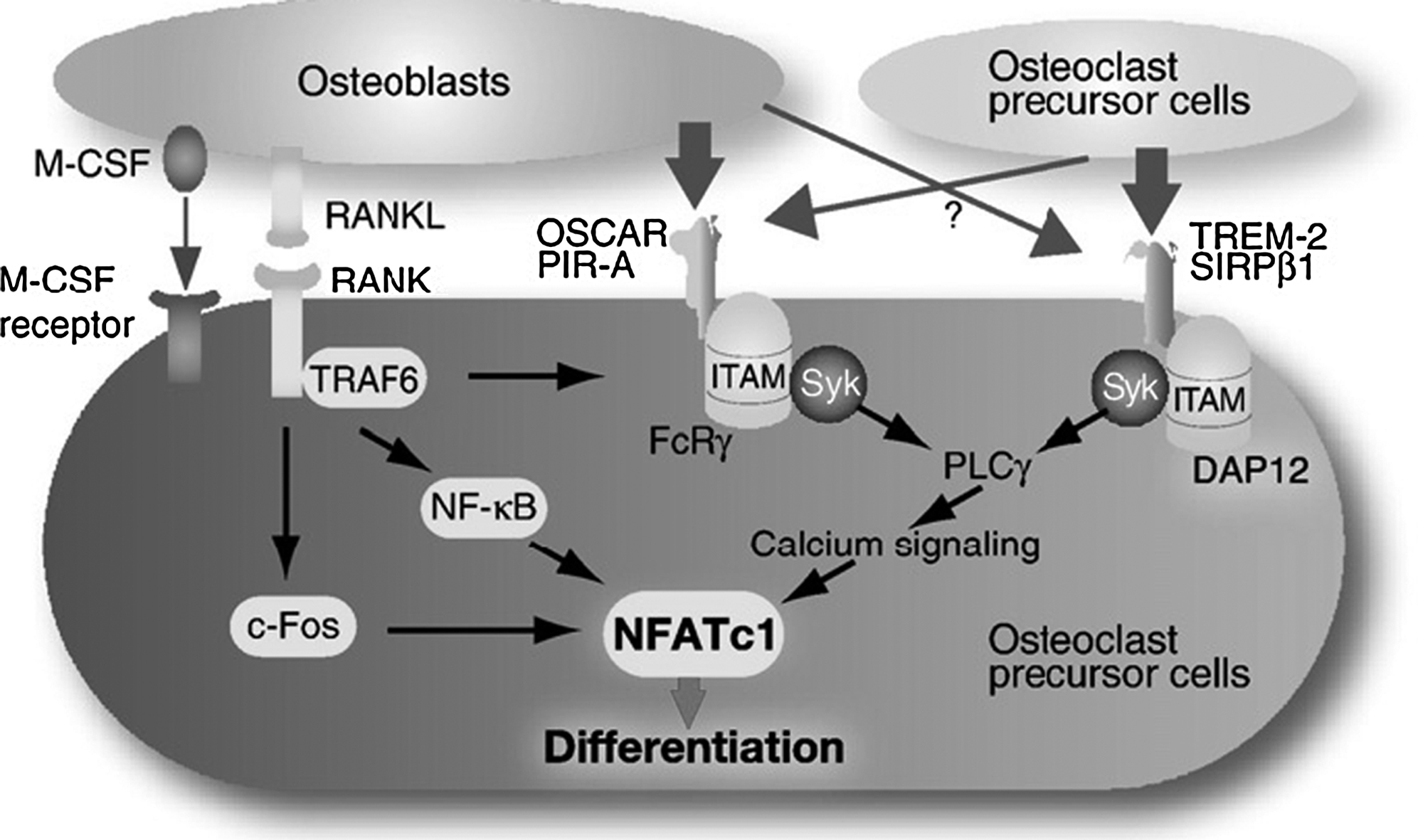
In addition to RANKL and M-CSF, osteoclast differentiation requires costimulatory signals mediated by receptor-associated adapter proteins containing immunoreceptor tyrosine-based activation motifs (ITAMs). Mice lacking two ITAM-bearing adapters, DNAX-activating protein (DAP)12 and the Fc receptor γ-chain (FcRγ) exhibit severe osteopetrosis due to failure of osteoclast differentiation. Phosphorylation of the ITAMs recruits Syk tyrosine kinase and in turn activate calcium signaling through phospholipase Cγ .
Osteoclasts derive from monocyte/macrophage precursors of the hematopoietic lineage. The Ets family transcription factor, PU.1, is critically important for the earliest events in osteoclastogenesis . PU.1-deficient mice lack not only osteoclasts, but also macrophages, while preserving the ability to produce early monocytic cells . PU.1 is thought to regulate the transcription of the M-CSF receptor, c-fms .
Two main transcription factor complexes, AP-1 and MITF1, are downstream of the M-CSF pathway in osteoclastogenesis. Mice lacking the AP-1 component, c-Fos, lack osteoclasts and are osteopetrotic but have an increased number of bone marrow macrophages . These data suggest that c-Fos has an important role in differentiation of hematopoietic precursors into osteoclasts rather than macrophages. Therefore c-Fos can be placed downstream of PU.1 in the pathway leading to a fully differentiated osteoclast. The microphthalmia gene product, MITF, is mutated in the mi/mi mouse, which is characterized by pigmentation (melanocyte) defects, mast cell defects, and osteopetrosis . MITF is essential for the fusion of mononuclear precursors into multinucleated osteoclasts and directly regulates genes important for osteoclast function such as those encoding TRAP, cathepsin K, and osteoclast-associated receptor (OSCAR) .
Downstream of RANK activation, TRAF6 is required for transduction of the RANK signal ( Fig. 3.6 ). TRAF6 interacts with the cytoplasmic tail of RANK, and deletion of this interaction domain abolishes RANK-mediated activation of NF-kB . That NF-kB is critical to osteoclastogenesis has been demonstrated by the finding that mice lacking the p50 and p52 subunits of NF-kB have osteopetrosis, a phenotype that can be rescued by bone marrow transplantation . c-Fos, a member of the AP-1 family mentioned earlier as downstream of PU.1, also serves an important function downstream of RANKL signaling . More recently, NFATc1 has been identified as a target of both TRAF6 and c-Fos pathways and may act as a major regulator of terminal osteoclast differentiation . Notably, a constitutively active form of NFATc1 in c-Fos null cells restores expression of osteoclast-specific genes, demonstrating that NFATc1 is a critical transcriptional regulator downstream of c-Fos during osteoclastogenesis . Activation of NFATc1 requires calcium signals that originate from phosphorylated ITAMs ( Fig. 3.6 ).
Related posts:
 The bone organ system: form and function
The bone organ system: form and function
 Impact of physical characteristics and lifestyle factors on bone density and fractures
Impact of physical characteristics and lifestyle factors on bone density and fractures
 Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
 Relationship between periodontal disease, tooth loss, and osteoporosis
Relationship between periodontal disease, tooth loss, and osteoporosis
 On the evolution and contemporary roles of bone remodeling
On the evolution and contemporary roles of bone remodeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


