Slow-flow malformations
Sturge-Weber, Klippel-Trenaunay, Proteus syndrome, cutis marmorata, Maffucci syndrome, Gorham-Stout, blue rubber bleb nevus syndrome, CLOVES (sometimes)
High flow
Bonnet-Dechaume-Blanc syndrome, Parkes Weber, Rendu-Osler, Cobb, Servelle-Martorell, CLOVES (sometimes)
Sturge-Weber Syndrome (SWS)
SWS, called also “encephalotrigeminal angiomatosis,” is a rare congenital disease characterized by port-wine stain of the face (in the forehead and upper eyelid, so-called VI area), glaucoma, mental retardation, and ipsilateral leptomeningeal vascular malformation.
The dermis of the V2 (maxillary) and V3 (mandibular) areas is made of cells from the mesencephalic neural crests, cells not forming leptomeninges: this is why SWS is not linked to a PWS occupying the V2 and V3 skin, without VI location [2] (Fig. 37.1).
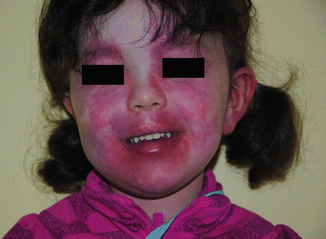
Fig. 37.1
Patient with Sturge-Weber syndrome and Port-wine stain involving the VI area. In this case the capillary malformation involves also the V2-V3 area and extends bilaterally
The syndrome is likely caused by a somatic mutation in the anterior neural primordium [2]. A recent work identified a somatic activating mutation in GNAQ, a gene that encodes a mediator of the G-protein-coupled receptor cascade [3].
The same mutation was identified also in non-syndromic cases (Parkes Weber syndrome). This implies that the origin of PWS and SWS is related. PWS might represent a late development of the somatic mutation in endothelial cells, while the SWS can be the result of an early mutation in progenitor cells that are precursors to a larger variety of cell types and tissues [3].
Epilepsy is the most severe symptom and may be devastating; it usually starts very early in life, between birth and 1 year. About 10 % of infants with VI PWS actually have leptomeningeal vascular anomalies. Cognitive deficit and mental retardation, loss of developmental milestones, and motor deficit contralateral to the meningeal lesions follow the onset of epilepsy, with a correlation between prolonged seizure and further severe developmental, motor, and intellectual deficits [3].
Prevention of the first seizure by prophylactic antiepileptic treatment aims at avoiding these severe developmental deficits. The seizures decrease strongly after puberty [3]. Ocular follow-up is also mandatory because of the risk of choroidal vascular anomaly and of glaucoma. The PWS can be treated by pulsed dye laser after pharmacological control of epilepsy [4, 5].
Klippel-Trenaunay Syndrome (KTS) and Related Syndromes
KTS, described in 1900 by the French neurologist Maurice Klippel and Paul Trenaunay [6], is a complex vascular malformation sited in the limbs which has a classical triad of signs: nevus, limb overgrowth, and dilated, abnormal superficial veins. These are the characteristics of the syndrome:
1.
Diffuse capillary malformation (CM) scattered over an extremity and adjacent trunk with apparently unsystematic spreading and nonassociated abnormality.
2.
Hypertrophy of the same extremity, with proportionate overgrowth over the years (angio-osteo-hypertrophy).
3.
A slow-flow combined and complex vascular malformation with venous (VM) and, very often, lymphatic malformations (LM), manifesting as either lymphedema or lymphatic vesicles (Fig. 37.2). KTS is associated with SWS in some patients [2].
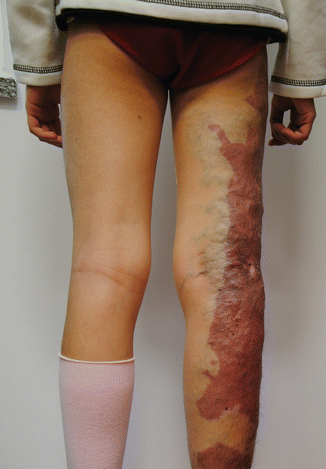
Fig. 37.2
Patient affected by Klippel-Trenaunay syndrome with Port-wine stain interesting the lateral aspect of the lower limb, associated superficial dysplastic varicose veins and mild hypertrophy of the involved limb
Vascular anomalies in the pelvis and also in the gut can be sometimes combined with the limb anomalies (see Chap. 46).
Even if association of lymphatic defects is considered a part of the syndrome, there are no accordance if, in the absence of LM, the case should be included or not in KTS.
Moreover, it remains unclear at which degree of combination of the above signs a case should be defined KTS or not. Klippel and Trenaunay described “incomplete cases” with only two of the signs of the triad. The reality is that today, many cases of vascular malformations of the limbs (and also sited in other areas!) are defined KTS.
To increase confusion, several cases in the literature are presented as “Klippel-Trenaunay-Weber syndrome” which can be defined a nonsense. If the main differences between KTS and PWS should be the presence/absence of venous and AV malformations (VM and no AVM in KTS, no VM and AVM in PWS), there should be no reason for this further combination. However, slight AV communications exist also in VM and may vary in extension.
Many years ago, the great pioneer in vascular malformations, Edmondo Malan, being aware of this confusionary condition about KTS and PWS, wrote: “Klippel-Trenaunay-Parkes-Weber syndrome is meaningless and should be abandoned” [7].
The diagnosis of KTS should clear precisely the existing defects by studying arterial, venous, and lymphatic system. The main tests are as follows:
US/Doppler duplex scan, MRI, phlebography, lymphoscintigraphy, and possible investigations for associated intestinal and urinary vascular anomalies [8, 9]. Anomalies of the deep veins of the limb (atresia, hypoplasia, or incompetence) can also be present and should be excluded (Fig. 37.3).
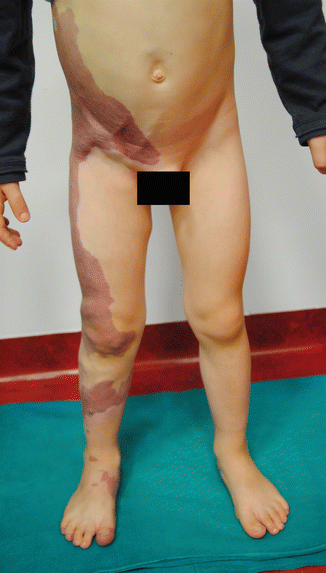
Fig. 37.3
Young patient affected by capillary malformation involving the lower limb and trunk associated to atresia of the ipsilateral common femoral vein. Compensatory dilated epigastric veins are visible on the lower part of the abdomen
Patients with KTS and limb hypertrophy are not at higher risk of Wilms’ tumor than the general population, as suspected before, and therefore, they do not need routine Wilms’ tumor screening unless the patient has generalized hemihypertrophy [10].
Monitoring of limb length difference progression is mandatory in pediatric patients by orthopedic assessment once a year. Epiphysiodesis is considered at around 10–13 years of age, depending on the growth curve of the child, when leg discrepancy is more than 2–3 cm and is increasing. Elastic garments are useful on a lifelong basis if the defects are not treated.
Early surgical treatment of varicose veins and dilated marginal vein is advisable in order to prevent chronic progressive distal venous hypertension [11]. Laser photocoagulation (FPDL or Nd:YAG laser) is useful for treatment of bleeding angiokeratomas and PWS [11] (Fig. 37.4).
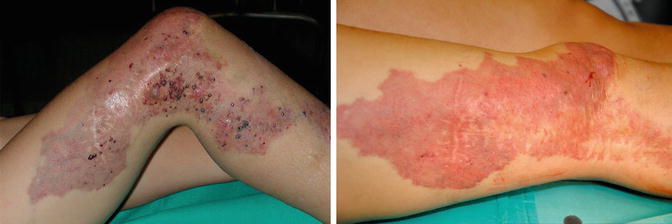
Fig. 37.4
Angiokeratoma Fig 37 B Result after laser treatment
Parkes Weber Syndrome (PWS)
PWS includes the same clinical “triad” as KTS: nevus, dilated superficial veins, and limb overgrowth [12]. However, two main differences are typical of PWS: absence of venous anomalies and presence of AV shunts, often multiple small-caliber AV shunts.
Also this syndrome, as KTS, is unclear in the limits as several questions are open. The main one is if all or only some of the cases with AV malformations of the limb should be defined PWS or not.
The therapy is similar to that of KTS with the exception of intervention on the venous vessels. Embolization procedures are not always possible because of the small calibers of the affected vessels.
Servelle-Martorell Syndrome
This syndrome is characterized by diffuse venous malformations and inhibition of limb bone growth resulting in limb length difference (angio-osteo-hypotrophy) [13]. The venous malformations are often extratruncular and infiltrate the limb till bones with local compression and thinning of the bone itself. Often diffuse phlebolythes are visible on x-ray examination. Deep main veins may be normal.
Treatment is based by occlusion of the diffuse venous defects with foam sclerosis or alcohol. However, treatment can be very long as malformations are infiltrating and extended. Sometimes, some limited painful areas can be resected surgically.
This defect should not be confused with Klippel-Trenaunay syndrome in which there is a limb overgrowth instead of shortening.
Cutis Marmorata
This malformation is characterized by the presence of multiple dilated capillaries and veins in the reticular dermis. Atrophic cutaneous ulcerations may be present. Asymmetries of the limbs are described in some cases. The main clinical features of the syndrome are the presence of reticulated erythema without response to heat exposure. There is no treatment for cutis marmorata as it is a benign condition that improves with age. In 50 % of the patients, the erythema disappears in the first 2 years of age [14–16].
Blue Rubber Bleb Nevus Syndrome
Blue rubber bleb nevus syndrome is a venous malformation with mainly limited vascular lesions affecting the skin, the mucosae, and the bowel. Skin lesions may be dark blue keratotic spots, blebs of normal skin color, or large venous or lymphatic masses. Bleeding from intestinal lesions may lead to anemia.
Complications of the syndrome are intussusception, volvulus, and mesenteric venous ischemia. Diagnosis is possible by CT scan; however, the best way to demonstrate the presence of this disease in the bowel is capsule endoscopy.
Cutaneous malformations may be treated successfully by surgical ablation or sclerotherapy. In case of stable disease because of less gut bleeding, iron and vitamin supplementations are sufficient. In case of severe intestinal bleeding, octreotides, rapamycin, and thalidomide are effective. Single-lesion, surgical excision is curative. Otherwise, bleeding control may be obtained by sclerotherapy and/or laser treatment in localizations which are achievable by endoscopic approach [17–20]. Intestinal location is discussed in detail in Chap. 46.
Proteus Syndrome
Proteus syndrome is characterized by cutaneous nevi, venous malformations, and swelling of the soft tissue with asymmetric overgrowth of bones. The overgrowth becomes evident in adolescential time.
The malformation is caused by a genetic mosaic pattern (activating mutation in AKT1 kinase), but there is no genetic transmission [21]. In some patients, some evidence of a PTEN gene anomaly was found.
The differential diagnosis is to the Klippel-Trenaunay syndrome and the hemihyperplasia lipomatosis syndrome. The criteria for the diagnosis of Proteus syndrome are summarized in Table 37.2 [22–24].
Table 37.2
Criteria for the diagnosis of Proteus syndrome
Group A | Connective tissue nevus |
Group B | Epidermal nevus |
Disproportionate area of overgrowth (one or more) | |
Bilateral anexial cystadenomas or parotid adenoma | |
Before the second decade of life | |
Group C | Lipomata or localized absence of fat |
Vascular malformations | |
Lung cysts | |
Facial malformations |
Gorham-Stout Syndrome
This syndrome, also called “disappearing bone syndrome,” was first described by Jackson in 1838 and successively published by Gorham and Stout in 1955 [25, 26]. It is typical of extreme osteolysis associated with lymphatic malformations. The cause of this disease is not known. Any bone may be involved, and in addition, pleural and abdominal effusions of the lymph and chyle are not uncommon. The pathological specimens of the involved bones show a hyperproliferation of lymphatic vessels which communicate with the blood vessels. The therapy is not well validated: some authors obtained good results using interferon and steroids, others injecting ethanol into the bone [27–29]. We had an excellent result with a Gorham-Stout case that has a large cyst on the distal femoral bone and extended smaller cysts along the whole bone. Repeated alcohol injections brought to a closure of the cyst and to a progressive strengthening of the whole bone which, at the beginning of treatment, was very easy to puncture.
Related posts:
 Hemangiomas of Infancy: Epidemiology
Epidemiology of Vascular Malformations
Hemangiomas of Infancy: Epidemiology
Epidemiology of Vascular Malformations
 Clinical Aspects in Vascular Malformations
Clinical Aspects in Vascular Malformations
 Imaging of Vascular Malformations
Imaging of Vascular Malformations
 Classification of Arteriovenous Malformation and Therapeutic Implication
Classification of Arteriovenous Malformation and Therapeutic Implication
 Vascular Malformations of the Limbs: Treatment of Venous and Arteriovenous Malformations
Vascular Malformations of the Limbs: Treatment of Venous and Arteriovenous Malformations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


