Several signaling systems downstream of G-CSFR have been identified that are defective or hyperactivated in myeloid cells of patients with congenital neutropenia: severely reduced expression of myeloid-specific transcription factors LEF-1 and C/EBPα, severely reduced expression and functions of HCLS1 protein, severely reduced expression of neutrophil elastase protein, dramatic compensatory up-regulation of the NAMPT/NAD + /SIRT pathway leading to continuous activation of emergency granulopoiesis via the transcription factor C/EBPβ, and hyperactivation of STAT5 protein by tyrosine phosphorylation.
Key points
- •
Severely reduced expression of myeloid-specific transcription factors, lymphoid enhancer binding factor 1 (LEF-1) and C/EBPα.
- •
Severely reduced expression and functions of HCLS1 protein.
- •
Severely reduced expression of neutrophil elastase (NE) protein.
- •
Dramatic compensatory up-regulation of the nicotinamide phosphoribosyltransferase (NAMPT)/NAD + /SIRT pathway, leading to continuous activation of emergency granulopoiesis via the transcription factor C/EBPβ.
- •
Hyperactivation of STAT5 protein by tyrosine phosphorylation.
Granulocyte colony-stimulating factor receptor signaling is severely affected in congenital neutropenia patients
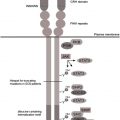
Granulocyte colony-stimulating factor (G-CSF) receptor (G-CSFR) activation with ligand binding induces myeloid cell proliferation, survival, and differentiation. Acquired somatic mutations within the CSF3R gene and/or defects in the CSF3R downstream signaling pathways abrogate myeloid differentiation and might lead to either leukemic transformation or congenital neutropenia (CN). In CN patients, the levels of G-CSF mRNA in mononuclear cells, the concentrations of biologically active G-CSF in serum, and the CSF3R expression in myeloid cells are considerably elevated compared with healthy individuals. Daily injections of pharmacologic doses of G-CSF (100–1000 times higher than physiologic levels), however, are needed to increase peripheral blood neutrophil counts to greater than 1000/μL in CN patients. Therefore, the authors assume that G-CSFR downstream signaling is severely defective in CN, leading to maturation arrest of granulopoiesis.
Granulocyte colony-stimulating factor receptor signaling is severely affected in congenital neutropenia patients
Granulocyte colony-stimulating factor (G-CSF) receptor (G-CSFR) activation with ligand binding induces myeloid cell proliferation, survival, and differentiation. Acquired somatic mutations within the CSF3R gene and/or defects in the CSF3R downstream signaling pathways abrogate myeloid differentiation and might lead to either leukemic transformation or congenital neutropenia (CN). In CN patients, the levels of G-CSF mRNA in mononuclear cells, the concentrations of biologically active G-CSF in serum, and the CSF3R expression in myeloid cells are considerably elevated compared with healthy individuals. Daily injections of pharmacologic doses of G-CSF (100–1000 times higher than physiologic levels), however, are needed to increase peripheral blood neutrophil counts to greater than 1000/μL in CN patients. Therefore, the authors assume that G-CSFR downstream signaling is severely defective in CN, leading to maturation arrest of granulopoiesis.
Common pathomechanism of defective granulopoiesis in congenital neutropenia patients harboring either ELANE or HAX1 mutations
Autosomal dominant mutations in the ELANE and autosomal recessive mutations in the HAX1 have been identified in a majority of CN patients (60% and 10%, respectively). Mutations in other genes, such as G6PC3 , GPT1 , TAZ1 , WAS , and so forth, are rare and are discussed in articles elsewhere in this issue by Boztug and colleagues. This article predominantly reports on data from patients harboring ELANE and HAX1 mutations. Ultimate defects in intracellular signaling pathways downstream of either ELANE or HAX1 mutations leading to the defective granulopoiesis were elusive until recently. Clinical observations revealed that CN patients harboring either ELANE or HAX1 mutations have comparable bone marrow (BM) morphology, responses to G-CSF therapy, and requirements of G-CSF dosages. Also the risk (approximately 20%) of developing leukemias is comparable in both patient subgroups. Based on these clinical data, the authors suggest a common pathomechanism of defective G-CSF–triggered granulopoiesis downstream of both mutated genes ( Fig. 1 ). In the past few years, the authors identified several novel signaling pathways that are activated by G-CSF in healthy individuals and are dramatically deregulated in hematopoietic cells of CN patients, leading to ineffective granulopoiesis.
A lack of LEF-1 and C/EBPα transcription factor expression in myeloid cells of congenital neutropenia patients
The authors aimed to identifying signaling pathways or related myeloid transcription factors that are severely reduced in CN patients causing abnormal granulopoiesis downstream of the ELANE or HAX1 mutations. mRNA expression profiles were compared between CD33 + BM myeloid cells (predominantly promyelocytes) of CN patients, healthy individuals, and patients with neutropenia other than CN (cyclic neutropenia, idiopathic neutropenia, or neutropenia due to metabolic defects, such as glycogenosis type Ib); all groups were treated or not with G-CSF. Severely diminished expression and functions of the transcription factors, LEF-1 and C/EBPα, in myeloid cells of CN patients were identified compared with all other studied groups. LEF-1 expression was abrogated in CN patients harboring either ELANE or HAX1 mutations, which suggested LEF-1 as a possible common candidate factor for defective G-CSF signaling in both groups of CN patients. The authors demonstrated markedly diminished G-CSF–triggered in vitro granulocytic differentiation of CD34 + cells of healthy individuals after knockdown of LEF-1. At the same time, LEF-1 rescue in hematopoietic cells of CN patients restored maturation arrest of granulopoiesis. Furthermore, LEF-1 was found to regulate granulopoiesis by direct binding to the gene promoter of the known granulocyte-specific transcription factor C/EBPα and activated expression of C/EBPα in myeloid cells ( Fig. 2 ). In CN patients, C/EBPα expression was also severely diminished, representing a possible reason for the defective granulocytic differentiation.
Imbalance in the transcriptional regulation of granulopoietic versus monopoietic differentiation programs of myeloid progenitor cells of congenital neutropenia patients
CN patients have elevated levels of peripheral blood monocytes, which can be explained by compensatory monocytosis due to diminished neutrophil counts and functions. Another reason for elevated production of monocytes and diminished granulopoiesis is deregulated expression of lineage-specific (granulocyte-specific and monocyte-specific) transcription factors in myeloid progenitor cells of CN patients. Proper regulation of granulopoiesis versus monopoiesis is tightly regulated by balanced expression of granulocyte-specific transcription factors (eg, C/EBPα) and monocyte-specific transcription factors (eg, PU.1). Thus, C/EBPα expression levels elevated above PU.1 levels lead to granulocytic differentiation, but prevalence of PU.1 expression above C/EBPα levels shifts differentiation toward monocytes. The authors found elevated levels of PU.1 expression and severe diminished levels of C/EBPα in myeloid progenitor cells of CN patients, which may be a reason for elevated monocyte production and defective granulopoiesis in these patients. Previously, it has been demonstrated that LEF-1 binds to the upstream regulatory element of the PU.1 gene promoter, inhibiting PU.1 expression. Thus, a lack of LEF-1 in myeloid cells of CN patients could cause up-regulation of PU.1 and down-regulation of C/EBPα expression, inducing a shift from granulocytopoiesis toward monocytopoiesis of myeloid progenitors in CN patients.
The authors and other investigators also demonstrated dose-dependent effects of LEF-1 in G-CSFR–triggered myelopoiesis: defective LEF-1 expression causes neutropenia, but overexpression of LEF-1 results in elevated proliferation of human hematopoietic cells and development of acute myeloid leukemia (AML) in a mouse model. Inhibition of LEF-1 by small hairpin RNA (shRNA) induces apoptosis and cell cycle arrest of the AML cell lines and primary AML blasts. LEF-1 belongs to the LEF-1/T-cell factor (TCF) family (TCF-1, TCF-3, and TCF-4) of high mobility group (HMG) domain transcription factors of the Wnt signaling pathway that recognize DNA consensus motifs through HMG box DNA-binding domain. LEF-1 does not have a transactivation domain but contains a DNA-binding domain. Therefore, it requires additional interaction partners with a transactivation domain to activate target genes. The best-known interaction partner of LEF-1 is β-catenin. The authors demonstrated, however, that activation of C/EBPα and subsequent stimulation of granulopoiesis by LEF-1 are β-catenin independent. LEF-1 lacking β-catenin binding domain was able to activate C/EBPα.
HCLS1, the hematopoietic-specific interaction partner of LEF-1, connects HAX1 mutations with diminished LEF-1 expression
The authors were interested in identifying hematopoiesis-specific interaction partners of LEF-1. Moreover, it was unclear why mutations in the ubiquitously expressed protein HAX1 induce isolated neutropenia and no defects in other tissues. The mechanism downstream of HAX1 mutations leading to defective LEF-1 expression was also unknown. The authors performed screening of candidate proteins with hematopoiesis-specific expression and activity, which can interact and regulate LEF-1 transcription factor, and found that hematopoietic cell-specific Lyn substrate 1 (HCLS1 or HS1) interacted with LEF-1 protein, transporting LEF-1 into the nucleus with G-CSF stimulation and subsequently inducing LEF-1 autoregulation, C/EBPα activation, and granulocytic differentiation. HCLS1 protein is expressed at high levels in human myeloid cells, is associated with Lyn and Syk, and is phosphorylated on stimulation with G-CSF. HAX1 is a HCLS1-associated protein X1 and, in CN patients with HAX1 mutations, the authors found profound defects in the G-CSF–triggered phosphorylation of HCLS1, which subsequently leads to abrogated nuclear transport of LEF-1, reduced autoregulation of LEF-1, and neutropenia. Therefore, the authors were able to identify a direct link between HAX1 mutations, defective LEF-1 expression, and isolated neutropenia in CN patients. Moreover, the authors identified one hematopoietic-specific interaction partner of LEF-1.
What are the main functions of HCLS1? HCLS1 belongs to the SRC homology 3 domain adapter proteins and can initiate activation of receptor-coupled tyrosine kinases. The proline-rich region is the site of tyrosine phosphorylation of HCLS1 and, therefore, responsible for many interactions with SH2-domain–containing proteins. The phosphorylation sites have been implicated in the regulation of the HCLS1 activity through the connection with diverse tyrosine kinases, such as Syk, Lyn, and Lck, and the adapter proteins, such as Grb2. Phosphorylation of HCLS1 on Tyr-397 by Syk and Lyn leads to HCLS1 translocation into the nucleus. Helix-turn-helix repeat and coiled-coil domains of HCLS1 are required for binding to F-actin and activation of the Arp2/3 complex. HCLS1 protein is also associated with PI3K/Akt pathway. Knowing the functions of the HCLS1 protein, the authors further analyzed if HCLS1 is involved in the G-CSF–triggered F-actin rearrangement. Previously, the authors demonstrated that F-actin assembly induced by G-CSF treatment is severely impaired in myeloid cells of CN patients. G-CSF treatment of CD34 + cells of healthy individuals led to a rapid and transient increase in F-actin content. As seen in CN patients, basal F-actin levels were significantly increased in CD34 + cells after knockdown of HAX1 or HCLS1. G-CSF was unable, however, to regulate the amount of F-actin in the absence of HAX1 or HCLS1. Therefore, the authors concluded that HCLS1 and HAX1 are important for G-CSF-induced F-actin assembly and that, in CN patients, mutations in the HAX1 with subsequent defects in HCLS1 activation may contribute to abnormal F-actin.
Because HCLS1 is also associated with PI3K/Akt pathway and PI3K/Akt is activated by G-CSF, the authors further analyzed if HCLS1 is involved in G-CSF–triggered activation of PI3K/Akt signaling and found that treatment of CD34 + cells with G-CSF led to phosphorylation of PI3K p85 (on Tyr458) and of Akt (on Ser473), which were both markedly reduced in cells transduced with HCLS1-specific or HAX1-specific shRNA. Similarly, much lower amounts of G-CSF–dependent phospho-PI3K p85 (Tyr458) and phospho-Akt (Ser473) were detected in CD34 + cells of patients with CN compared with cells from healthy individuals. Thus, the authors identified a new essential player in the G-CSFR signaling pathway that is involved in nuclear transport and activation of LEF-1 transcription factor, activation of PI3K/Akt pathway, and deregulation of F-actin assembly. Similar to LEF-1, the functions of HCLS1 are dose dependent ( Fig. 3 ). Thus, the authors also detected dramatically elevated HCLS1 protein levels in blasts of AML patients. A large cohort of AML patients also has insertion in the HCLS1 gene, which is localized in the proline-rich region of the HCLS1 protein. This insertion has been described as transmitting an accelerated B-cell receptor signaling in B lymphocytes inducing receptor-independent activation. Similar hyperactivating events may be responsible for hyperproliferation of AML blasts.
Nicotinamide phosphoribosyltransferase–dependent compensatory hyperactivation of emergency granulopoiesis in congenital neutropenia patients
The authors were further interested in how G-CSF treatment can overcome maturation arrest of granulopoiesis in CN patients despite the absence of LEF-1 and C/EBPα in myeloid cells. The hypothesis was that for responses to physiologic dosages of G-CSF, additional compensatory mechanisms, which are independent of LEF-1 and C/EBPα, should be activated with G-CSF treatment in CN patients. In the search for such mechanisms, the authors identified NAMPT as an essential enzyme mediating G-CSF–triggered granulopoiesis in healthy individuals and in CN patients. Treatment of healthy individuals with G-CSF resulted in up-regulation of NAMPT levels in myeloid cells via STAT3 and subsequent release of NAMPT into the plasma. At the same time, intracellular NAMPT and NAD + amounts in myeloid cells, as well as plasma NAMPT and NAD + levels, were even more dramatically elevated by G-CSF treatment of individuals with CN. The molecular events triggered by NAMPT included elevation of NAD + , NAD + -dependent activation of protein deacetylase sirtuin-1 (SIRT1), binding of SIRT1 to the myeloid-specific transcription factors, C/EBPα and C/EBPβ, and subsequent activation of these transcription factors ( Fig. 4 ). G-CSF and G-CSFR are target genes of C/EBPs and treatment of myeloid cells with NAMPT induced autocrine C/EBP-triggered production of G-CSF and expression of G-CSFR, providing a feedback loop of G-CSF:G-CSFR signaling. C/EBPα transcription factor regulates steady-state granulopoiesis and, in cases of bacterial infections or situations that require rapid granulocyte production, emergency granulopoiesis triggered by C/EBPβ is activated. In patients with CN, expression of C/EBPα transcription factor is severely diminished due to a lack of LEF-1 ; therefore steady-state granulopoiesis could not be activated. G-CSF treatment, however, induces expression of C/EBPβ transcription factor in these patients via NAMPT and SIRT1 and operates via emergency arm of granulopoietic signaling. Because, for emergency granulopoiesis, continuous presence of the strong activation stimulus is required, G-CSF should be administered daily in high therapeutic doses to reach sufficient granulocyte numbers in CN patients ( Fig. 5 ). Thus, another novel player in G-CSF signal transduction pathway has been identified and an important role of deacetylation of myeloid-specific transcription factors in G-CSFR–triggered myeloid differentiation has been established. Modulation of protein deacetylation by treatment of neutropenia patients with nicotinamide (vitamin B 3 ), which is converted into NAD + by the enzyme NAMPT, could be applied.
Related posts:
 Genetics and Pathophysiology of Severe Congenital Neutropenia Syndromes Unrelated to Neutrophil Elastase
Genetics and Pathophysiology of Severe Congenital Neutropenia Syndromes Unrelated to Neutrophil Elastase
 Granulocyte Colony-Stimulating Factor Receptor Signaling
Chronic Granulomatous Disease
Granulocyte Colony-Stimulating Factor Receptor Signaling
Defective G-CSFR Signaling Pathways in Congenital Neutropenia
Leukocyte Adhesion Deficiencies
Granulocyte Colony-Stimulating Factor Receptor Signaling
Chronic Granulomatous Disease
Granulocyte Colony-Stimulating Factor Receptor Signaling
Defective G-CSFR Signaling Pathways in Congenital Neutropenia
Leukocyte Adhesion Deficiencies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


