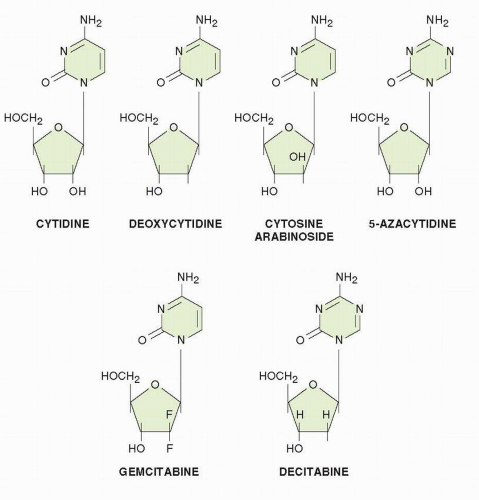
Ara-C is one of the most effective agents in the treatment of acute myelogenous leukemia (AML)
3 and is incorporated into virtually all standard induction regimens for this disease, generally in combination with an anthracycline (daunorubicin hydrochloride or idarubicin hydrochloride). Ara-C is also a component of consolidation and maintenance regimens in AML after remission is attained.
4 High-dose ara-C confers particular benefit in AML patients with certain cytogenetic abnormalities related to the core binding factor that regulates hematopoiesis (t8:21, inv 16, del 16, t16:16).
5 Ara-C is also active against other hematologic malignancies, including Burkitt’s lymphoma,
6 acute lymphocytic leukemia,
7 and chronic myelogenous leukemia
8 but has little value as a single agent against solid tumors. This limited spectrum of activity has been attributed to the lack of metabolic activation of this agent in solid tumors and its selective action against rapidly dividing cells. The essential features of ara-C pharmacology are described in
Table 10-1.
Mechanism of Action
Ara-C acts as an analogue of deoxycytidine (CdR) and has multiple effects on DNA synthesis. Ara-C undergoes phosphorylation to form arabinosylcytosine triphosphate (ara-CTP), which competitively inhibits DNA polymerase
α in opposition to the normal substrate deoxycytidine 5′-triphosphate (dCTP).
9 This competitive inhibition has been demonstrated with crude DNA polymerase from calf thymus
9 and with purified enzyme from human leukemic cells
10 and from murine tumors.
11 Ara-CTP has an affinity for human leukemia cell DNA polymerase
α in the range of 1 × 10
−6 mol/L and competes with dCTP. In intact cells, its effects are antagonized by the addition of CdR, the precursor of dCTP.
12 Ara-CTP inhibits DNA polymerase
β with lesser potency.
13 The effects of ara-C on DNA polymerase activity extend not only to semiconservative DNA replication but also to DNA repair. Repair of ultraviolet light damage to DNA, a function of polymerase
α, is blocked more potently than the repair of photon-induced or
γ radiation-induced strand breaks,
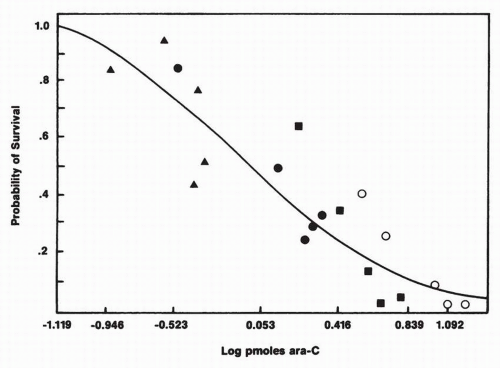
14 which are repaired by a different polymerase. In addition to its inhibition of DNA synthesis, however, it becomes incorporated into DNA, a feature that correlates closely with cytotoxicity
15,
16 (
Fig. 10-2). In fact, a preponderance of evidence suggests that this is the major cytotoxic lesion in ara-C-treated cells. Drugs that prevent ara-C incorporation into DNA, such as aphidicolin, block its cytotoxicity.
17 In cell culture experiments, a linear relationship exists between picomoles of ara-C incorporated into DNA and the log of cell survival for a wide range of drug concentrations and durations of exposure. Thus, drug toxicity is a direct function of incorporation into DNA, and the latter varies directly with drug concentration and duration of exposure.
18 Once incorporated into DNA, ara-C is excised slowly,
19 and the incorporated ara-C inhibits template function and chain elongation.
17,
20,
21 In experiments with purified enzyme and calf thymus DNA, the consecutive incorporation of two ara-C or two arabinosyl-5-azacytidine (ara-5-aza-C) residues effectively stops chain elongation.
10 At high concentrations of ara-C, one finds a greater than expected proportion of ara-C residues at the 3′-terminus, a finding that implies potent chain termination.
19 These observations support the hypothesis that ara-C incorporation into DNA is a prerequisite for drug action and is responsible for cytotoxicity.
Ara-C also causes an unusual reiteration of DNA segments.
22 Human lymphocytes exposed to ara-C in culture synthesize small reduplicated segments of DNA, which results in multiple copies of limited portions of DNA. These reduplicated segments increase the possibility of recombination, crossover, and gene amplification; gaps and breaks are observed in karyotype preparations after ara-C treatment. The same mechanism, reiteration of DNA synthesis after its inhibition by an antimetabolite, may explain the high frequency of gene reduplication induced by methotrexate, 5-fluorouracil, and hydroxyurea (see relevant chapters). In summary, although ara-C has multiple effects on DNA synthesis, the most important antitumor mechanism seems to be its incorporation into DNA.
Other biochemical actions of ara-C have been described, including a relatively weak inhibition of ribonucleotide reductase
23 and formation of arabinosylcytosine diphosphate (ara-CDP)-choline. The latter functions as an analog of cytidine 5′-diphosphocholine (CDP-choline) and inhibits synthesis of membrane glycoproteins and glycolipids.
24 Ara-C also has the interesting property of promoting differentiation of leukemic cells in tissue culture, an effect that is accompanied by decreased c-
myc oncogene expression.
25 These changes in morphology and oncogene expression occur at concentrations above the threshold for cytotoxicity and may simply represent terminal injury of cells. After exposure to ara-C, both normal and malignant cells undergo apoptosis in experimental models.
26The mechanism by which ara-C induces apoptosis is uncertain. It may be triggered by p53 in response to DNA breaks (see above) and by futile attempts to excise incorporated ara-C nucleotides. Ara-C stimulates the formation of ceramide, a potent inducer of apoptosis.
27 Ara-C induces production of diacylglycerol, which activates protein kinase C (PKC), a response that opposes apoptosis in hematopoietic cells. The lethal actions of ara-C may depend, at least partially, on its relative effects on the PKC and ceramide pathways.
Cells exposed to ara-C display marked alterations in transcription factors. Ara-C induces AP-1 (a dimer of jun-fos or jun-jun proteins) and NF-kB, and induction has been temporally associated with apoptosis.
28,
29 PKC inhibitors promote ara-C-induced apoptosis despite their antagonizing c-
jun up-regulation, calling into question the involvement of c-
jun expression in apoptosis secondary to ara-C.
30 Ara-C induces pRb phosphatase activity, another possible mechanism responsible for p53-independent G
1 arrest and apoptosis.
31 The resulting hypophosphorylated pRb binds to and inactivates the E2F transcription factor, which inhibits the transcription of genes responsible for cell-cycle progression.
32
Cellular Pharmacology and Metabolism
Ara-C penetrates cells by a carrier-mediated process shared by physiologic nucleosides.
33 Several different classes of transporters for nucleosides have been identified in mammalian cells; the most extensively characterized in human tumors is hENT1, the equilibrative transporter, identified by its binding to nitrobenzylthioinosine (NBMPR). The number of transport sites on the cell membrane is greater in AML cells than in acute lymphocytic leukemia cells and can be enumerated by incubation of cells with NBMPR.
34 The hENT1 transporter is highly upregulated in biphenotypic leukemia associated with the 11q23 MLL gene (4:11) translocation.
35 Uptake occurs rapidly. A steady-state level of intracellular drug is achieved within 90 seconds at 37°C.
36,
37 Studies of Wiley et al.
33 and others
34,
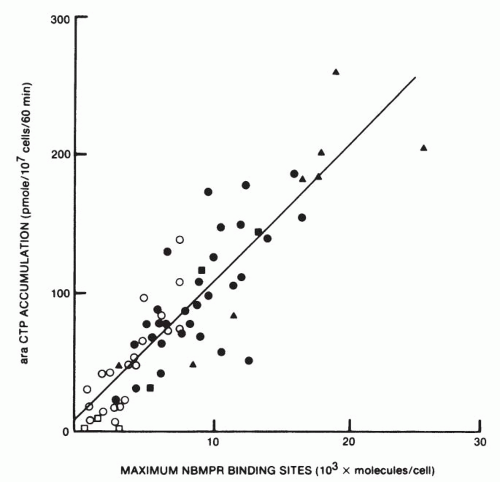
37 suggest that the NBMPR transporter plays a limiting role in the action of this agent in that the formation of the ultimate toxic metabolite ara-CTP is strongly correlated with the number of transporter sites on leukemic cells (
Fig. 10-3). A single point
mutation in the hENT1 carrier confers resistance in leukemic cell lines in vitro.
36 At drug concentrations above 10 μmol/L, the transport process becomes saturated, and further entry takes place by passive diffusion.
37 hENT1 is strongly inhibited by various receptor tyrosine kinase inhibitors, an interaction that could limit ara-C use with targeted drugs.
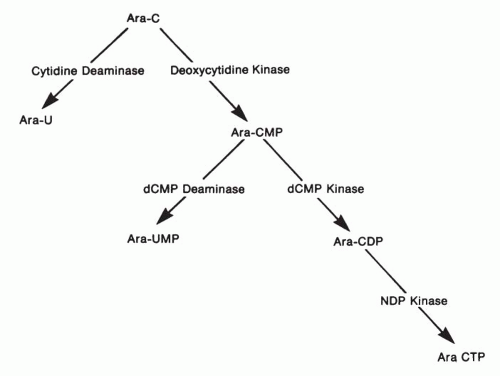
38As shown in
Figure 10-4, ara-C must be converted to its active form, ara-CTP, through the sequential action of three enzymes: (a) CdR kinase, (b) deoxycytidine monophosphate (dCMP) kinase, and (c) nucleoside diphosphate (NDP) kinase. Ara-C is subject to degradation by cytidine deaminase, forming the inactive product uracil arabinoside (ara-U); arabinosylcytosine monophosphate (ara-CMP) is likewise degraded by a second enzyme, dCMP deaminase, to the inactive arabinosyluracil monophosphate (ara-UMP). Each of these enzymes, with the exception of NDP kinase, has been examined in detail because of its possible relevance to ara-C resistance.
The first activating enzyme, CdR kinase, is found in lowest concentration (
Table 10-2) and is believed to be rate limiting in the process of ara-CTP formation. The enzyme is a 30.5-kDa protein that phosphorylates ara-C and many pyrimidine and purine nucleosides and their analogs. The gene coding for CdR kinase and a mutated version found in ara-C resistant cells have been cloned.
39,
40 The rate-limiting role of CdR kinase in ara-C activation is illustrated by transfection of malignant cell lines with retroviral vectors containing CdR kinase cDNA, a maneuver that substantially increases the susceptibility of cells to ara-C, gemcitabine, and purine analogues.
41 Ara-C cytotoxicity increases when intracerebral gliomas in rats are transduced with CdR kinase.
42CdR kinase activity is highest during the S phase of the cell cycle.
43 The
Km, or affinity constant, for ara-C is 20 μmol/L, compared with the higher affinity or 7.8 μmol/L for the physiologic substrate CdR.
44 This enzyme is strongly inhibited by dCTP but weakly inhibited by ara-CTP. This lack of “feedback” inhibition allows accumulation of the ara-C nucleotide to higher concentrations. PKC-
α, the activity of which is increased after ara-C exposure, has been implicated in phosphorylation and activation of CdR kinase. This observation raises the possibility that ara-C at high doses may potentiate its own metabolism by induction of the PKC activator, diacylglycerol.
45The second activating enzyme, dCMP kinase,
46 is found in several hundred-fold higher concentration than CdR kinase. Its affinity for ara-CMP is low (
Km = 680 μmol/L) but greater than the affinity for the competitive physiologic substrate dCMP. Because of its relatively poor affinity for ara-CMP, this enzyme could become rate limiting at low ara-CMP concentrations. The third activating enzyme, the diphosphate kinase, appears not to be rate limiting because it is present in very high concentration and the intracellular pool of ara-CDP is only a fraction of the ara-CTP pool.
47Opposing the activation pathway are two deaminases found in high concentration in some tumor cells as well as in normal tissues. Cytidine deaminase is widely distributed in mammalian tissues, including intestinal mucosa, liver, and granulocytes.
48 It is found in granulocyte precursors and in leukemic myeloblasts in lower concentrations than in mature granulocytes, but even in these immature cells, the deaminase level exceeds the activity of CdR kinase, the initial activating enzyme.
44,
48 The second degradative enzyme, dCMP deaminase (
Fig. 10-4), regulates the flow of physiologic nucleotides from the dCMP pool into the deoxyuridine monophosphate (dUMP) pool from which point dUMP is ultimately converted to deoxythymidine 5′-phosphate (dTMP) by thymidylate synthase.
49 The enzyme dCMP deaminase is strongly activated by intracellular dCTP (
Km = 0.2 μmol/L) and strongly inhibited by deoxythymidine triphosphate in concentrations of 0.2 μmol/L or greater. Ara-CTP weakly activates this enzyme (
Km = 40 μmol/L
50 and, thus, would not promote degradation of its own precursor nucleotide, ara-CMP. The affinity of dCMP deaminase for ara-CMP is somewhat higher than the affinity of dCMP kinase for the same substrate, but the activity of these competitive enzymes depends greatly on their degree of activation or inhibition by regulatory triphosphates (dCTP), and dCMP deaminase concentration in leukemic myeloblasts is slightly less than that of dCMP kinase (
Table 10-2).
The balance between activating and degrading enzymes, thus, is crucial in determining the quantity of drug converted to the active intermediate, ara-CTP. This enzymatic balance varies greatly among cell types.
44 CdR kinase activity is higher and cytidine deaminase activity lower in lymphoid leukemia than in acute myeloblastic leukemia. Enzyme activities vary also with cell maturity; deaminase increases dramatically with maturation of granulocyte precursors, whereas kinase activity decreases correspondingly.
48 Thus, admixture of normal granulocyte precursors with leukemic cells in human bone marrow samples complicates the interpretation of enzyme measurements unless normal and leukemic cells are separated. In general, cytidine deaminase (D) activity greatly exceeds kinase (K). The kinase:deaminase ratio averages 0.03 in human AML, whereas the enzyme activities are approximately equal in acute lymphoblastic leukemia and Burkitt’s lymphoma. Thus, the biochemical setting seems to favor drug activation by lymphoblastic leukemia cells if these initial enzymes play a rate-limiting role.
In fact, this may not be the case. Chou et al.
47 found that human AML cells formed 12.8 ng of ara-CTP per 10
6 cells after 45 minutes of incubation with 1 × 10
−5 mol/L ara-C. Acute lymphoblastic leukemia cells formed less ara-CTP, 6.3 ng/10
6 cells, and as expected, the more mature chronic myelocytic and chronic lymphocytic leukemia cells formed lesser amounts of ara-CTP (4.7 to 5.2 ng/10
6 cells). The likelihood is that other factors, such as transport across the cell membrane and regulatory effects of intracellular nucleoside triphosphate concentrations, may limit ara-CTP formation.
In addition to its activation to ara-CTP, ara-C is converted intracellularly to ara-CDP-choline,
51 an analog of the physiologic CDP-choline lipid precursor. However, ara-C does not inhibit incorporation of choline into phospholipids of normal or transformed hamster embryo fibroblasts.
24 Ara-CMP does inhibit the transfer of galactose,
N-acetylglucosamine, and sialic acid to cell surface glycoproteins. Further, high concentrations (approaching 1 mM) of ara-CTP inhibit the synthesis of cytidine monophosphate (CMP)-acetylneuraminic acid, an essential substrate in sialylation of glycoproteins.
52 Thus, ara-C treatment could alter membrane structure, antigenicity, and function.
Biochemical Determinants of Cytosine Arabinoside Resistance
The foregoing consideration of ara-C metabolism and transport makes it clear that a number of factors could affect ara-C response. Not surprisingly, many of these factors have been implicated in various preclinical models of ara-C resistance. The most frequent abnormality found in resistant leukemic cells recovered from mice treated with ara-C has been decreased activity of CdR kinase.
53 In cultured cells exposed to a mutagen and then to low concentrations of ara-C, some single-step mutants developed high-level resistance to ara-C through loss of activity of CdR kinase, whereas other resistant clones exhibited markedly expanded dCTP pools, presumably through increased cytidine-5′-triphosphate (CTP) synthetase activity or through deficiency of dCMP deaminase.
54,
55 As mentioned previously, specific mutations and deletions in the gene coding for CdR kinase derived from resistant cells have been described.
39,
40The role of cytidine deaminase in experimental models of resistance is less clear. Retrovirus-mediated transfer of the cytidine deaminase cDNA into 3T3 murine fibroblast cells significantly increases drug resistance to ara-C and other analogs such as 5-aza- 2′-CdR and gemcitabine. This phenotype of increased cytidine deaminase activity and drug resistance is reversed by the cytidine deaminase inhibitor tetrahydrouridine (THU).
56 Other genes, including proto-oncogenes, may affect ara-C response. Transfection of rodent fibroblasts and human mammary HBL 100 cells with
c-H-ras conferred resistance to ara-C, an event attributed to decreased activity of CdR kinase.
57 On the other hand,
N-ras or
K-ras mutations strongly correlated with increased ara-C sensitivity in the screening of human tumor cell lines from the National Cancer Institute’s in vitro drug screen.
58 Ras mutations are found in 20% of AML cases, and these patients appear to derive greatest benefit from high-dose ara-C regimens.
59 Although various molecular lesions have been implicated as causing ara-C resistance in animals, their relevance to resistance in human leukemia is less certain. Clinical studies have described specific biochemical changes in drug-resistant cells from patients with leukemia, including deletion of CdR kinase,
60 increased cytidine deaminase,
61 a decreased number of nucleoside transport sites,
62 and increased dCTP pools.
63 Other clinical investigators have not been able to correlate resistance with either CdR kinase or cytidine deaminase or their ratio.
64,
65 All studies have shown extreme variability in enzyme levels among patients with leukemia. Thus, no agreement exists as to the specific changes responsible for resistance in human leukemia.
Although
specific biochemical lesions associated with resistance in humans are unclear, the current understanding of ara-C action suggests that the ultimate formation of ara-CTP and the duration of its persistence in leukemic cells determine response.
47,
66 Chou et al.
47 found greater ara-CTP formation in leukemic cells of responders when these cells were incubated in vitro with ara-C, but even this correlation was not confirmed in other studies.
67,
68,
69Preisler et al.
70 found that the duration of remission induced by ara-C-containing regimens was strongly correlated with the ability of cells to
retain ara-CTP in vitro after removal of ara-C from
the medium. Attempts to monitor ara-CTP formation in leukemic cells taken from patients during therapy have not disclosed useful correlations of ara-CTP levels or intracellular persistence with response.
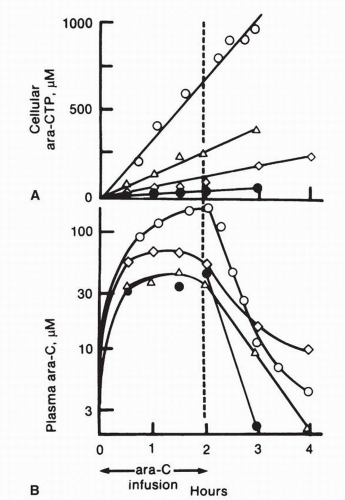
69,
71 Ara-CTP has an intracellular half-life of about 3 to 4 hours. Again, considerable variability has been observed in the rates of formation of ara-CTP, and this rate does not correlate well with plasma ara-C pharmacokinetics in individual patients (
Fig. 10-5).
The cellular response to ara-C-mediated DNA damage also governs whether the genotoxic insult results in cell death. Ara-C incorporation into DNA stalls the replication fork for cells in active DNA synthesis, activating ATR and Chk 1, checkpoint kinases that block cell cycle progression and allow for removal of ara-C from DNA. Absence of either of these checkpoints sensitizes cells to apoptosis. Levels of expression of apoptotic proteins influence response. Overexpression of the antiapoptotic proteins Bcl-2 and Bcl-X
L in leukemic blasts causes in vitro resistance to ara-C-mediated apoptosis.
72 The intracellular metabolism of ara-C and its initial effects on DNA are not modified by Bcl-2 expression, which suggests that Bcl-2 primarily regulates the more distal steps in the ara-C-induced cell death pathway. Although the precise mechanism by which these proteins prevent ara-C-induced cytotoxicity remains to be elucidated, Bcl-2 and Bcl-X
L have been shown to antagonize ara-C-mediated cell death by caspase activation.
72 The fact that antisense oligonucleotides directed against Bcl-2 increase the susceptibility of leukemic blasts to ara-C-induced apoptosis in vitro,
73 and that patients whose blasts express high levels of Bcl-2 respond poorly to ara-C-containing regimens,
74 further suggests the pivotal role of Bcl-2 in ara-C resistance.
Phosphorylation of apoptotic or DNA damage response factors may also determine the outcome of ara-C exposure. Phosphorylation of Bcl-2 is required for its antiapoptotic function, and a functional role for PKC-
α in Bcl-2 phosphorylation and suppression of apoptosis has been postulated,
75 although this observation has not been confirmed by others.
76 Attempts at pharmacological inhibition of PKC-
α activity have increased ara-CTP formation but had variable effects on cytotoxicity in culture, possibly due to their simultaneous activation of BcL-2.
76 Altered phosphorylation of transcription factors also influences the cellular response to ara-C toxic insult. Ara-C-induced activation of PKC and mitogen-activated protein kinase (MAPK) increases c-
jun expression and phosphorylation,
27,
77 and hyperphosphorylation of the AP-1 transcription factor has been associated with ara-C resistance in human myeloid leukemic cell lines in vitro.
78Clinical studies of determinants of ara-C response are complicated by the fact that ara-C is almost always given in combination with an anthracycline or an anthraquinone. Thus, a complete response or long remission duration does not necessarily imply sensitivity to ara-C. A lack of response does imply resistance to both agents in the combination, except for the not-infrequent cases in which failure can be attributed to infection or inability to administer full dosages of drug. With these limitations, the duration of complete response is probably the most appropriate and most important single yardstick of drug sensitivity because it reflects the fractional cell kill during induction therapy, but no single factor has emerged as a determinant of remission duration.
Cell Kinetics and Cytosine Arabinoside Cytotoxicity
In addition to biochemical factors that determine response, cell kinetic properties exert an important influence on the results of ara-C treatment. As an inhibitor of DNA synthesis, ara-C has its greatest cytotoxic effects during the S phase of the cell cycle perhaps because of the requirement for its incorporation into DNA and the greater activity of anabolic enzymes during S phase. The duration of exposure of cells to ara-C is directly correlated with cell kill because the longer exposure period allows ara-C to be incorporated into the DNA of a greater percentage of cells as they pass through S phase. The cytotoxic action of ara-C is not only cell-cycle phasedependent but is influenced by the rate of DNA synthesis. That is, cell kill in tissue culture is greatest if cells are exposed during periods of maximal rates of DNA synthesis, as in the recovery period after exposure to a cytotoxic agent. In experimental situations, it has been possible to schedule sequential doses of ara-C to coincide with the peak in recovery of DNA synthesis and thus to improve the therapeutic results.
79In humans, the influence of tumor cell kinetics on response is unclear. Although, earlier studies showed that the complete remission rate seems to be
higher in patients who have a high percentage of cells in S phase,
80 remissions are
longer in patients with leukemias that have long cell-cycle time.
81
Alternate Schedules of Administration
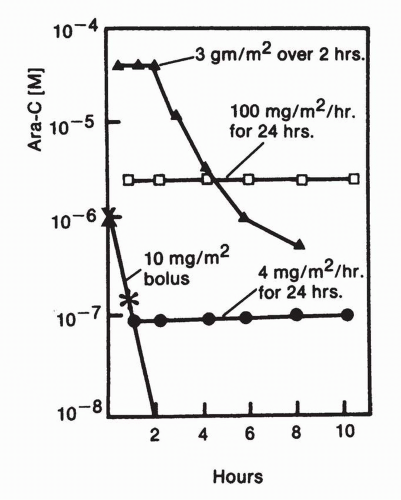
Although ara-C is used most commonly in regimens of 100 to 200 mg/m
2/d for 7 days, other high-dose and low-dose schedules have been used in treating leukemia. The more effective of these newer regimens have been high-dose schemes, usually 2 to 3 g/m
2 every 12 hours for six doses.
97 High-dose ara-C is used primarily in the consolidation phase for acute myelocytic leukemia.
4 The rationale for the higher-dose regimen initially rested on the assumption that ara-C phosphorylation is the rate-limiting intracellular step in the drug’s activation and could be promoted by raising intracellular concentrations to the
Km of CdR kinase for ara-C, or approximately 20 μmol/L. Above this level, further increases in ara-C do not lead to increased ara-CTP because the phosphorylation pathways enzymes become saturated.
98Others have examined the clinical activity of low-dose ara-C, particularly in older patients with myelodysplastic syndromes.
99 These regimens have used dosages in the range of 3 to 20 mg/m
2/d for up to 3 weeks, with the expectation that low doses would produce less toxicity and promote leukemic cell differentiation (or apoptosis). The persistence of chromosomal markers for the leukemic cell line in remission granulocytes has been documented, findings that support induction of differentiation.
100 In general, although the low-dose regimens produce less toxicity, myelosuppression often supervenes and less than 20% of patients achieve meaningful improvement in blood counts.