INTRODUCTION
SUMMARY
Cutaneous T-cell lymphoma (CTCL) is a heterogeneous group of malignant lymphomas that share the propensity for malignant T lymphocytes expressing cutaneous lymphocyte antigen to infiltrate the skin. Mycosis fungoides (MF) is the most common variant of CTCL, representing approximately 50 percent of all cases. Sézary syndrome (SS) is a leukemic variant of MF, affecting approximately 5 percent of patients with MF.1 MF and SS are the most common malignant proliferations of mature memory T lymphocytes of the helper phenotype (CD4+CD45RO+),2 which renders patients immunocompromised even at the earliest stages of the disease. Advanced stages are associated with severe immune suppression. Diagnosis is established by skin biopsy, followed by staging work up which includes radiologic imaging and pathologic evaluation of the lymph nodes, internal organs, blood, and marrow, as appropriate, according to presenting manifestations of the disease.
MF is divided into early and advanced stages for therapeutic and prognostic reasons. In early stages, the disease follows an indolent course and has a favorable prognosis. In advanced stages, the prognosis is poor. There are numerous therapeutic options. No treatment has been definitively proven to improve survival, but newer studies suggest that survival is longer than historically documented.3,4 Multiagent chemotherapy is not a useful option because it is inferior to immune modulating and biologic therapies.5 Considering the overall protracted course of the disease, its indolent character, immunocompromised status of the patients, and absence of definitive therapy, aggressive multiagent chemotherapy contributing to immunosuppression should be reserved for end-stage palliation or as a bridge to stem cell transplantation with the goal of definitive cure.6,7,8 There are several FDA-approved single agents for therapy of MF, which are safe and effective against the disease and may be used in the therapeutic ladder prior to multi-agent regimens.9,10,11 Several single agents and combinations of new and older agents are in clinical trials now to test for efficacy in MF and SS. Because no single therapy is considered to be the standard of care for MF and SS, clinical trials remain a viable option for most patients. The goal of therapy is to induce long-term remissions without compromising patients’ immunity and improvement of the quality of life.
Acronyms and Abbreviations
ALCL, anaplastic large cell lymphoma; ALK-1, anaplastic lymphoma kinase-1; ATRA all-trans retinoic acid; BCNU, bis-chloroethyl-nitrosourea or carmustine; CLA, cutaneous lymphocyte antigen; CT, computed tomography; CTCL, cutaneous T-cell lymphoma; DD, denileukin diftitox; ECP, extracorporeal photopheresis; EORTC, European Organisation for Research and Treatment of Cancer; HDACi, histone deacetylase inhibitor; HTLV-1, human T-lymphotropic virus type 1; Ig, immunoglobulin; IL, interleukin; LEBT, localized electron beam therapy; LyP, lymphomatoid papulosis; MF, mycosis fungoides; MMAE, monomethyl auristatin E; NBUVB, narrow band UVB; NCCN, National Comprehensive Cancer Network; NK, natural killer; PCALCL, primary cutaneous anaplastic large cell lymphoma; PCR, polymerase chain reaction; PET, positron emission tomography; PUVA, psoralen with UVA; RXR, retinoid X receptor; SDT, skin-directed therapy; SS, Sézary syndrome; TCR, T-cell receptor; Th2, T-helper type 2; TNMB, tumor, node, metastasis, blood; TSEBT, total-skin electron beam therapy; UV, ultraviolet; UVA, ultraviolet A; UVB, ultraviolet B; WHO, World Health Organization.
DEFINITION AND HISTORY
In 1806, Baron Jean-Louis Alibert described a patient who presented with skin patches that grew into plaques and mushroom-like tumors and first coined the term mycosis fungoides (MF).12 In 1938, Sézary and Bouvrain described a syndrome of pruritus, generalized exfoliative erythroderma, and abnormal hyperconvoluted lymphoid cells in the blood.13 Today this condition is referred to as Sézary syndrome (SS), a condition seen in a subset of patients with MF.
Prior to the 1970s, cutaneous lymphomas were believed to be cutaneous counterparts of the systemic lymphomas. In 1975, Lutzner and associates14 suggested the term cutaneous T-cell lymphoma (CTCL), recognizing that despite the significant similarities of malignant cell morphology and phenotype to systemic T-cell lymphomas, the cutaneous lymphomas represented distinct entities. Although this definition has helped to distinguish cutaneous lymphomas from systemic disease, it has also led to inappropriately using the umbrella term CTCL interchangeably with MF. Common World Health Organization (WHO)–European Organisation for Research and Treatment of Cancer (EORTC) classification was first developed in 200515 to resolve differences between various classifications (Table 103–1) and is currently accepted as the standard for CTCL staging and classification.16
|
EPIDEMIOLOGY
MF is twice as common in males as in females. The median age at diagnosis is 55 years. Americans of African descent have a higher incidence of MF and a poorer prognosis than Americans of European descent. MF occurs least often in Asians and Hispanics. Evidence for a genetic predisposition (germline transmission of susceptibility) in patients with CTCL is inconclusive. An approximately 6 percent increase in incidence of CTCL per decade was documented from the early 1970s to 1998, but the incidence subsequently leveled off and stabilized, with a current incidence of approximately one per 100,000.17 Incidence is highly age-dependent, with the highest incidence of 3.6 per 100,000 of adults after the age of 70 years. Approximately 3000 new cases of MF are reported annually, comprising approximately 3 percent of all lymphomas. The mortality rate varies widely according to the stage of disease. Stage I mortality does not differ from the mortality of age-matched controls. However, stage IV patients have a 27 percent 5-year survival and 10 percent 15-year survival. Median survival of patients with SS is 1.5 years. The mortality rate of MF in the United States has been declining, possibly because of earlier diagnosis of the disease.1,18
ETIOLOGY AND PATHOGENESIS
The etiologies of MF and SS are unknown, although epidemiologic features are suggestive of an infectious origin, including a predilection for elderly individuals and a higher-than-expected incidence in immune-suppressed patients.19 However, studies to date have failed to reveal consistent associations between any particular infectious agent and CTCL, including novel infectious agents.20 A “persistent antigen stimulation” hypothesis was proposed as an initial event after MF was observed to be a disease of mature CD4+ memory T cells, but the stimulating antigen is not known.21,22 MF also may be viewed as a disease of immune dysregulation. Tumor progression is associated with decreased antigen-specific T-cell responses and impaired cell-mediated cytotoxicity. On the other hand, improved survival is associated with intact cell-mediated immunity. Progression of MF is associated with progressive T-helper type 2 (Th2) skewing and increased production of Th2 cytokines. This alteration accounts for many of the immune abnormalities associated with advanced MF, such as hypereosinophilia, increased serum immunoglobulin (Ig) A and IgE levels, impaired natural killer (NK) cell function, and impaired cellular immunity.23,24 Late-stage MF and SS are associated with declining immunocompetence,25 resulting in severe life-threatening infections, and a high incidence of secondary malignancies. The latter increase is not attributable to prior treatment with carcinogenic agents alone.26 Fifty percent of deaths among patients with MF result from infections.
Environmental factors have been suggested in the etiology of CTCL in Europe,27,28 but have not been confirmed by epidemiologic studies in the United States.
CLINICAL FEATURES
The clinical presentation of MF is highly variable. Cutaneous manifestations of the disease result from skin infiltration by malignant cutaneous lymphocyte antigen (CLA)-positive lymphocytes and depend on the extent of skin involvement. Patients initially may present with “chronic dermatitis” that is resistant to therapy, which is often misdiagnosed as spongiotic dermatitis (so-called eczema), “psoriasis-like dermatitis,” or other chronic, nonspecific dermatoses, usually associated with pruritus. Histologically, diagnosis may be difficult, especially in the early stages of the disease and in its erythrodermic form, as the abnormal atypical infiltrate can be minimal and can be masked by normal inflammatory infiltrates in the skin, or it can be misinterpreted as a normal inflammatory infiltrate because of its mature CD4+ phenotype.
MF may progress through distinct stages of skin involvement, ranging from patch (Fig. 103–1A) to plaque (Fig. 103–1B) to tumor (Fig. 103–1C), but any type of lesion may progress or lesions may arise de novo. For descriptive purposes, the skin manifestations of MF are divided into patch stage (patch-only disease), plaque stage (both patches and plaques), and tumor stage (more than one tumor present, usually in the context of patches and plaques) (Fig. 103–1C). A patch is defined as a flat lesion with various degrees of erythema and fine scaling; it may be atrophic or poikilodermatous (Fig. 103–1D). A plaque is a well-demarcated erythematous, brownish, or violaceous lesion of at least 1 mm elevation with a variable amount of scale. Tumors are elevated at least 10 mm above the skin surface and may resemble a plaque or be dome shaped without significant scaling.
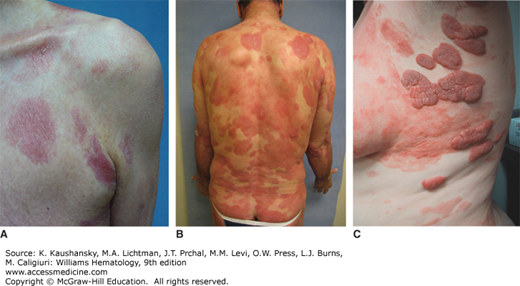
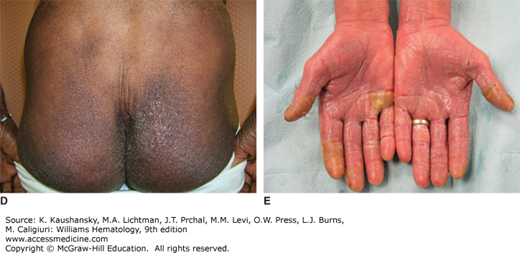
Distribution of the lesions depends on the clinical stage at presentation. In earlier stages, the lesions have a predilection for folds and non–sun-exposed body areas (“bathing trunk” distribution). In later stages, the lesions can affect the face, including development of ectropion, and other areas, such as palms and soles (keratoderma; see Fig. 103–1E). Tumors may be generalized, and ulceration is common. Progression through the stages is variable but commonly occurs over several years.29 Lesions usually are associated with pruritus, which may range from mild to excruciatingly severe, leading to insomnia, weight loss, depression, and suicidal ideation. Pruritus is one of the most important quality-of-life issues for these patients.30
Erythrodermic skin involvement occurs in 5 percent of patients with MF. Manifestations range from very faint to severe, with significant scaling, keratoderma, painful fissures of the hands and feet, nail dystrophy, and nail loss leading to the patient’s inability to walk and maintain daily activities. Severely inflamed skin serves as a breeding ground for bacteria and other pathogens, with resulting fevers, chills, and septicemia.19,31 Peripheral edema of the extremities may be significant in the later stages and lead to cardiovascular compromise.
Depending on the stage of presentation, patients may present with nodal and/or blood involvement and/or visceral metastases. The earliest stages of MF (e.g., Stage IA and B), may pursue a waxing and waning course, with minimal symptoms and may not have any negative prognostic implications for a patient. The more advanced stages usually, but not inevitably, present with symptoms of the disease. The symptomatology usually reflects the site and severity of involvement and ranges from completely asymptomatic to severe pain, organ malfunction, or at the end stage disease, multi-organ failure.
LABORATORY FINDINGS
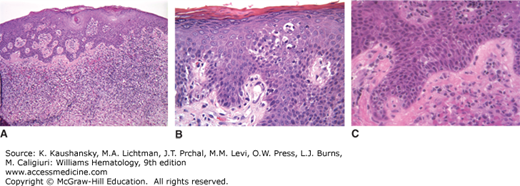
There is no definitive marker for MF or SS. The diagnosis of CTCL usually is established by correlating clinical and pathologic findings. Early lesions may show polymorphic infiltration (containing mixed inflammatory cells) compatible with several benign dermatoses. Classically, MF lesions show superficial band-like (lichenoid) lymphocytic infiltrate (Fig. 103–2A). The lymphocytes may range from small to large, with characteristic convoluted (cerebriform) nuclei. The hallmark of the malignant infiltrate in MF is epidermotropism (presence of lymphocytes in the epidermis without spongiosis) with formation of the epidermal clusters of lymphocytes around Langerhans cells termed Pautrier microabscesses (Fig. 103–2B). Atypical lymphocytes line up along the dermoepidermal junction and are surrounded by a halo artifact (Fig. 103–2C), which is an important feature of early disease.32 Superficial dermal collagen may be thickened, so called ropey collagen. In more advanced stages, the infiltrate is less polymorphic, with a predominance of larger atypical cells extending deeper into the dermis; epidermotropism may be lost.33 Transformation to large T-cell lymphoma (CD30+ or CD30−) may occur and carries a poor prognosis in the setting of MF.34
Figure 103–2.
Mycosis fungoides. Skin biopsies stained with hematoxylin and eosin. A. Lichenoid (band-like) lymphocytic infiltrate in the superficial dermis. B. Epidermotropic atypical lymphocytes lining the dermoepidermal junction in the absence of spongiosis-forming Pautrier microabscesses. Note, halo artifact around lymphocytes at the dermoepidermal junction. C. Atypical lymphocytes in the epidermis.
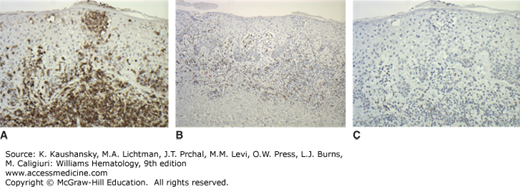
Immunophenotyping plays an important role in diagnosis. The cells usually are CD3+CD4+CD45RO+CD8−, a phenotype associated with mature helper-inducer T lymphocytes. Loss of maturation markers, such as CD7 and CD26 expression on CD4+ are important markers for malignant T lymphocytes.35,36 The cells may express T-cell activation markers, such as HLA-DR or CD25 (interleukin [IL]-2 receptor). Clonal rearrangement of the T-cell receptor (TCR) Vβ gene can be identified in approximately 90 percent of advanced cases of MF, but in only 50 percent of early stage cases.37 In rare instances, the classic clinical presentation of MF may be associated with an aberrant CD4 phenotype or may have CD4−dyCD8+ T-cell phenotype38,39 (Fig. 103–3A to C). Recent molecular studies have identified several new markers, which may prove useful as positive markers of the disease, including thymocyte selection-associated high mobility group box factor (TOX), plastin (PLS3), and killer cell immunoglobulin-like receptor KIR3DL2.40,41,42,43 Cytogenetic abnormalities are not consistently identified, but loss of heterozygosity on 10q and microsatellite instability may be seen in advanced-stage disease.44 A possible association exists with homozygous deletion of PTEN and CDKN2A and tumor-suppressor genes on 10p and 9p chromosomes, respectively. These may be silenced with progression of disease.45,46
MF is classified according to the widely accepted modified tumor, node, metastasis, blood (TNMB) classification, originally adopted in 1975 by the Mycosis Fungoides Cooperative Study Group47,48 and revised to match modern developments in the field.16 Accurate determination of the stage in MF and SS is of utmost importance because of its prognostic significance and critical role in selecting therapy. Cutaneous lesions are classified using the T staging system (Table 103–2). The area of the skin and type of the lesions were found to correlate with patient survival and are important prognostic predictors and need to be calculated at every visit to assess disease status.49 Prognosis varies according to tumor burden. The presence of tumors (T3) may indicate a worse prognosis than erythroderma (T4).18
|
The extent of extracutaneous disease usually correlates with the extent of skin involvement. In early disease, significant involvement of lymph nodes and blood is unlikely. However, lymphadenopathy is present in more than half of patients as disease advances and increases with progressive cutaneous involvement. Lymph nodes are assigned N category in the TNMB staging of MF16 (see Table 103–2). Computed tomography (CT) and positron emission tomography (PET) scans are used to assess involvement of lymph nodes.50,51 Excisional lymph node biopsy is usually recommended to assess the extent of the disease and nodal architecture, but other techniques, including fine-needle aspiration, are selectively used as well.52
Histopathologic examination of affected lymph nodes may show partial or complete effacement of normal architecture, with a monomorphic infiltrate of MF cells. However, in most cases, the nodal architecture is not effaced, and dermatopathic changes are present with varying numbers of atypical lymphocytes in the T-cell paracortical areas of the node. Even the presence of dermatopathic changes alone in the lymph nodes carries prognostic significance (see Tables 103–2 and 103–3).53,54 Abnormal lymph nodes should be biopsied regardless of the T stage.
| T | N | M | B | |
|---|---|---|---|---|
| IA | 1 | 0 | 0 | 0, 1 |
| IB | 2 | 0 | 0 | 0, 1 |
| IIA | 1, 2 | 1, 2 | 0 | 0, 1 |
| IIB | 3 | 0–2 | 0 | 0, 1 |
| III | 4 | 0–2 | 0 | 0, 1 |
| IIIA | 4 | 0–2 | 0 | 0 |
| IIIB | 4 | 0–2 | 0 | 1 |
| IVA1 | 1–4 | 0–2 | 0 | 2 |
| IVA2 | 1–4 | 3 | 0 | 0–2 |
| IVB | 1–4 | 0–3 | 1 | 0–2 |
Metastatic disease (including patients with positive nodes) is the most significant prognostic predictor (see Tables 103–2 and 103–3). Patients with visceral involvement that includes liver, spleen, pleura, and lung have a median survival of less than 1 year.55 Blood involvement may be an important predictor of progression and survival (Figs. 103–4 and 103–5).56 The number of circulating Sézary cells increases with advancing disease, and the cells are particularly prominent in patients with generalized erythroderma. However, even in early stage disease, a high frequency of clonal T cells in the blood may be detected using a highly sensitive polymerase chain reaction (PCR) technique, suggesting that early systemic disease is common.57 Blood involvement is rated as B category in the TNMB staging (see Table 103–2). For staging purposes, the B2 blood rating is equivalent to nodal involvement.58,59 The B2 rating is defined as (1) a Sézary cell count of 1000 cells/mm3 or more; (2) a CD4-to-CD8 ratio of 10 or higher caused by an increase in circulating T cells and/or an aberrant loss or expression of pan–T-cell markers by flow cytometry; (3) increased lymphocyte counts with evidence of a T-cell clone in the blood determined by the Southern blot or PCR technique; or (4) a chromosomally abnormal T-cell clone. Malignant cells also can be detected using sensitive techniques such as cytogenetics or TCR gene rearrangement studies.60,61,62,63,64 Patients with blood involvement have a higher likelihood of lymphadenopathy and visceral involvement. Marrow infiltration is infrequently detected by biopsy despite circulating malignant cells; it is identified at autopsy in 30 to 40 percent of cases. The cytologic appearance of the malignant cells in visceral organs is similar to that of the malignant cells in the skin.65
Figure 103–4.
Blood lymphocytes. A. Normal small lymphocyte. B. Sézary cell. Note the nuclear swirls and the light microscopic appearance of the Sézary cell nucleus. Without careful inspection in cases of lymphocytosis, Sézary cells can be mistaken for small lymphocytes as seen in chronic lymphocytic leukemia. C. Blood lymphocytes from a patient with mycosis fungoides and disseminated disease involving marrow and blood. Note clefted appearance of the nucleus. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
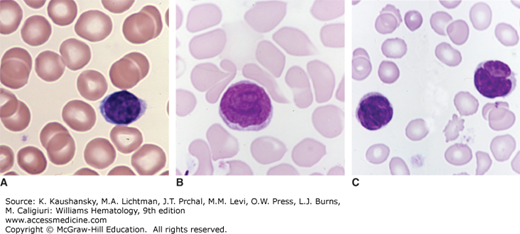
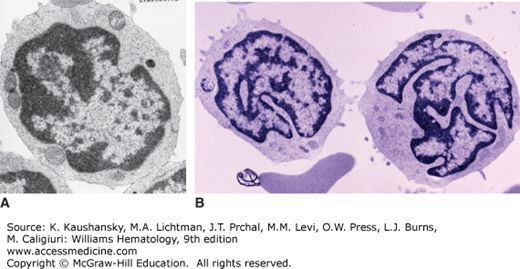
Figure 103–5.
Transmission electron micrographs of lymphocytes. A. Normal lymphocyte. B. Two lymphocytes from a patient with Sézary cells in the blood. The latter have the striking cerebriform nuclear abnormalities characteristic of Sézary cells. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
In the erythrodermic subset of MF, three T4 subsets can be identified (Table 103–4). In general, in SS, a triad of exfoliative erythroderma, generalized lymphadenopathy, and leukemia, has the worst prognosis among the forms of MF.
| Erythrodermic Subset (T4) | Preexisting MF | Blood |
|---|---|---|
| Sézary syndrome | Rarely | Leukemia: B2 |
Erythrodermic mycosis Fungoides | Always | Normal or minimally abnormal: B0–B1 |
| Erythrodermic cutaneous T-cell lymphoma, not otherwise specified | Absent | Normal or minimally abnormal: B0–B1 |
DIFFERENTIAL DIAGNOSIS
Diagnosis of MF is based on a constellation of findings, which include clinical presentation, skin and lymph node biopsies (if indicated), and blood evaluation. A number of benign dermatoses can mimic MF or SS, and may even have TCR gene rearrangements.66,67,68,69 Such benign conditions include psoriasis and psoriasiform dermatoses (such as pityriasis rubra pilaris, seborrheic dermatitis, contact dermatitis, and eczema), intertrigo, tinea, drug eruptions, and other conditions.
Cutaneous and systemic lymphomas other than MF should be considered in the differential diagnosis. Smoldering adult T-cell leukemia/lymphoma has a number of clinical features similar to MF, but it usually can be distinguished by the presence of antibodies to human T-lymphotropic virus type 1 (HTLV-1) and by other associated findings unusual in MF. However, this distinction may be difficult.70,71
Pagetoid reticulosis (Woringer-Kolopp disease)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


