Exposure to ultraviolet radiation
Ultraviolet A
Ultraviolet B
Therapy with methoxsalen and ultraviolet A radiation
Exposure to ionizing radiation
Genodermatosis
Oculocutaneous albinism
Xeroderma pigmentosum
Infection with human papillomavirus, especially types 6, 11, 16, and 18
Exposure to chemical carcinogens
Arsenic
Polycyclic aromatic hydrocarbons
Immunosuppression
Organ transplantation
Leukemia and lymphoma
Immunosuppressive medications
Chronically injured or diseased skin
Ulcers
Sinus tracts
Osteomyelitis
Radiation dermatitis
Certain chronic inflammatory disorders, such as dystrophic epidermolysis bullosa
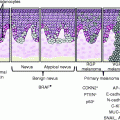
Precursor lesions
Actinic keratoses
Arsenical keratoses
Radiation-induced keratoses
Bowen’s disease (squamous cell carcinoma in situ)
Erythroplasia of Queyrat (squamous cell carcinoma in situ of the penis)
Albinism, xeroderma pigmentosum, and epidermodysplasia verruciformis are the cutaneous genetically inherited skin diseases with high propensity of risk for developing cSCC [15, 22, 23].
Immunodepression is related to high incidence of cSCC, that is up to 64–250-fold greater than that in the general population [26, 27]. In solid organ transplant patients, cSCC appears as multiple lesions, with high propensity of recurrence and metastatization [28]. In these patients, cSCCs develop within 10 years from organ transplant [29]. HPV has also been found in cSCC with higher evidence in immunosuppressed patients [20, 21]. cSCC could also occur in cutaneous sites with scars, sinus tracts, and burns. The latter are more aggressive with an overall metastatic rate of 40 % [16, 21].
Molecular Pathology of cSCC
Different pathways are deregulated in cSCC, dependently from etiological factor.
UV-Driven cSCC
UV radiations are very toxic agents for skin. The wave lengths of absorption depends upon molecular structure, being shorter around 200 nm for conjugated bonds in organic molecules and longer around 300 nm, in the range of solar spectrum, for linear repeats or ring structures. Therefore, DNA containing many ring structures, i.e., bases, and conjugated bonds represents the most relevant absorber of UV in the cell with consequent gene mutations [30]. The effects of UV on human skin can be considered acute or chronic. Between the acute effects, DNA damage is essentially due to the induction of primarily cyclobutane-type pyrimidine dimers and pyrimidine pyrimidone photoproducts [31–33].
Therefore, base modifications occur where a tandem of pyrimidine exists. Radiation induces primarily C → T and CC → TT transitions that are the hallmark of UV-induced mutagenesis [34]. Subsequently, when DNA polymerase cannot interpret the DNA template, the enzyme inserts A residues by default [35]. In addition, UV induces also cytosine photohydrates, purine photoproducts, and single-strand breaks in the DNA and the production of specific reactive oxygen species (ROS), responsible for the induction of single-strand breaks, DNA–protein cross-links, and altered bases in DNA [36, 37]. Acute UV-induced skin damage is also responsible for the induction of p53 expression through different mechanisms. p53 is a critical protein that causes cell cycle arrest, either favoring the cellular repair pathways to remove DNA damage before DNA synthesis and mitosis [38–40] or promoting apoptosis in cells with excessive DNA damage [41]. p53 protein increases in epidermal cells after exposure to UV radiation, as demonstrated in SKH-hr1 mice [42]. In details, it seems to be induced when DNA damage has characterized by pyrimidine dimers and DNA stands break [42]. In addition, p53 phosphorylation is induced by UV radiation through involvement of the ATM, ATR, and p38, and of the ERK1/2 and JNK-1 mitogen-activated protein (MAP) kinases-dependent pathways [43–46]. Thus, serine residue phosphorylation promotes stabilization of p53 by (1) disrupting the interaction of p53 with Mdm2, (2) enhancing p53 transcriptional activity, and (3) favoring p53 nuclear localization. The final effects of p53 activation are essentially cell cycle arrest followed by DNA repair occurring through two major pathways: non-excision repair (NER) and base excision repair (BER). In details, in NER pathway p53 transactivates the two xeroderma pigmentosum-associated gene products (p48XPE and XPC) and GADD45; [47–49] in BER pathway, p53 plays a role in regulating DNA polymerase, which is directly involved in DNA repair/synthesis [50]. When DNA damage is too deep, activated p53 in damaged skin induces apoptosis, through activation of a wide variety of transcriptional factors related to activation of the apoptosomal complex, such as the Bcl-2 family members Bax, p53-upregulated modulator of apoptosis (PUMA), Noxa, p53-upregulated apoptosis-inducing protein 1 (p53AIP1), and PIGa (galectin-7) [51–53]. Moreover, it has been demonstrated that UVB-induced apoptosis is related also to activation of Fas–Fas ligand, potent activators of pro-caspase 8 cleavage and, at the same time, of the cytochrome c release from mitochondria, followed by activation of the Apaf-1–pro-caspase 9 complex that both induce apoptosis [54]. Finally, UV-induced acute skin damage can also stimulate cell survival mechanisms and induce cell proliferation in the form of cell hyperplasia. In fact, UV triggers proliferation through activation of growth factor receptors such as erbB receptors and cytokine receptors, such as receptors for tumor necrosis factor (TNF) and interleukin-1 (IL1) [55, 56].
Chronic UV irradiation causes p53 mutation and loss of Fas–Fas ligand interaction with consequent apoptosis deregulation.
p53 mutations seem to be an early genetic event in the development of UV-induced skin cancers. In fact it has been described in approximately 53 % of premalignant actinic keratosis (AK) lesions [57]. In addition, 40 % of Bowen’s disease carry p53 mutations [58]. Many reports have recorded p53-mutant cell clones in nonneoplastic cells of sun-exposed skin [57–62]. However, p53 mutations in cSCC and in their respective precursors seem to be different from those in nonneoplastic skin areas [61]. This suggests that not all UV-induced p53mutations confer a malignant phenotype to neoplastic cells and that there is a latency from the occurrence of p53 mutation and cancer development. Some in vivo experiments have demonstrated that the occurrence of the most critical p53 mutations is recorded from 17 to 80 daily UVB exposures and skin tumors around 80 to 30 weeks later [63, 64]. Analyzing both C → T and CC → TT transitions in p53 gene, it has been observed that mutations are much earlier, being present at the first week from the beginning of chronic UV irradiation, with higher frequency at 4–8 weeks [65].
Therefore, selective expansion of p53-mutant cells occurs in the context of epidermis, due to both the resistance to UV-induced apoptosis and the proliferative advantage over normal keratinocytes in response to UV irradiation [64, 66, 67] (Fig. 3.1). Regression of a part of precancerous p53-positive clones can be observed after discontinuation from UV irradiation [64, 68] although tumor occurrence is only delayed [69].
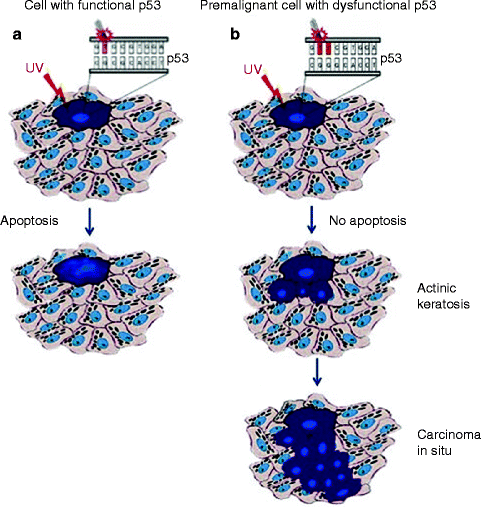
Fig. 3.1
(a) UV skin damage-induced apoptosis in functional p53 expressing normal skin; (b) Identification of p53-mutated clones in UV-induced cSCC
p53 Family Protein p63 and cSCC Pathogenesis
The epidermis is continuously regenerated by mitotically active keratinocytes in the inner basal layer which, following detachment from the basement membrane, migrates to the outer cornified layer (terminally differentiated compartment): a process called cornification. The epidermis is continuously renewed by a regulated balance between proliferation and differentiation. The renewal capacity is due to the presence of epidermal stem cells and transient amplifying (TA) cells in the basal layer of the interfollicular epidermis and in the bulge of the hair follicle. The importance of the transcription factor p63 for the formation of the epidermis and other stratified epithelia arises from two lines of evidence. First, p63 is strongly expressed in the innermost basal layer where epithelial cells with high clonogenic and proliferative capacity reside. Second, mice lacking all p63 isoforms have no epidermis or squamous epithelia (prostate, urothelium), probably due to a premature proliferative rundown of the stem and TA cells. Mice knockout for p63 (p63−/−) also show lack of epithelial appendages, such as mammary, salivary, and lachrymal glands, hair follicles, and teeth, and have truncated limbs and abnormal craniofacial development. This is most likely due to failure to maintain or differentiate the apical ectodermal ridge, a structure that is important for coordination of epithelial–mesenchymal interactions and is required for limb outgrowth and palatal and facial structure formation. p63 is expressed from two different promoters that generate two classes of proteins, TAp63, which contains the N-terminal transactivation(TA) domain, and the N-terminal truncated (DNp63) isoform, which lacks the transactivation domain. In addition, alternative splicing at the 3′ end of the transcripts generates three different C-termini: α, β, and γ. However, information about the protein expression levels of splice variants is not currently available due to the lack of isoform-specific antibodies, thus hampering the identification of specific functions.
It is emerging that p63 is involved in tumorigenesis and in controlling chemosensitivity.
The notion that the transcription factor p63 is essential for the formation of the epidermis and other stratified epithelia arises from the fact that mice lacking p63 show profound defects information of the epidermis. Remarkably, genetic mutations of p63 in man are causatively linked to ectodermal dysplastic syndromes. The involvement of p63 in the generation of skin cancer is related to its ability to control apoptosis through both extrinsic and intrinsic pathway. The loss of p63 in cancer can favor the survival of mutated cell clones that in the presence of p63 have not the possibility to develop towards cancer. Moreover, p63 is involved in the control of cell proliferation through the regulation of both the expression and activity of several growth factor receptors. In fact, p63 can affect the fibroblast growth factor receptor 2b (FGFR2b) receptor and its associated signaling, since it has been shown that there is a reduction in keratinocyte proliferation in the epidermis of Fgfr2b−/− embryos. In addition, the proliferative capacity of p63−/− mice keratinocytes could be also reduced as a consequence of increased expression of p21, a direct transcriptional repression target of DNp63. p63 functions as a selective modulator of Notch1-dependent transcription and function and also directly transactivates the Notch ligand, JAGG-1. Thus, a complex crosstalk between Notch and p63 could regulate the balance between keratinocyte self-renewal and differentiation. Moreover, a microarray analysis for p63 target genes, which was then validated in the genetically complemented mice, indicates that the function of p63 in epithelial development is at least in part mediated by IKKα (IκB kinase-α) and GATA-3 [70].
Fas and Fas Ligand
After the initial upregulation of Fas and Fas ligand expression induced by the acute exposure to UV, transcriptional inhibition of Fas ligand expression has been observed after 1 week of continuous UV irradiation in mice, with corresponding decrease in the number of apoptotic cells [65, 71]. Deregulation of Fas–Fas ligand-mediated signaling favors accumulation of p53 mutations, since Fas ligand-deficient mice with prolonged UV irradiation develop more p53 mutations than wild-type mice [54].
Growth Factor Receptors Belonging to the Superfamily of Erb
The tyrosine kinases human epidermal receptor (HER) family (epidermal growth factor receptor (EGFR), HER-2, HER-3, and HER-4) are transmembrane glycoproteins related to cell proliferation, differentiation, and apoptosis. Altered expression of the HER family is associated with several epithelial tumors such as breast carcinoma and esophageal squamous cell carcinoma. Small studies have also shown altered HER expression in localized squamous cell carcinoma when compared to normal skin. Given the paucity of unresectable or metastatic cSCC, reliable information on the frequency of EGFR expression is limited. One study of 13 metastatic specimens by IHC demonstrated that all had strong membranous expression of EGFR. Another study of locally advanced and nodally metastatic cutaneous SCC demonstrated EGFR expression above background in only 9 of 21 (43 %) specimens using a quantitative Western blotting technique. Another study using IHC and fluorescence in situ hybridization demonstrated higher levels of EGFR protein expression in cutaneous SCC than in the precursor actinic keratoses and an association between this elevated protein expression and higher EGFR gene copy number. One study, examining the role of EGFR in cSCC arising in the head and neck, demonstrated that primary lesions associated with subsequent metastases were more likely to overexpress EGFR (79 %) than those not associated with subsequent metastases (36 %). Interestingly, metastatic nodal disease exhibited only a 47 % rate of EGFR overexpression; EGFR overexpression in that study was not associated with EGFR gene amplification. Recently, a study on 55 primary tumors and 22 lymph node metastases derived from cSCC of the trunk and extremities has demonstrated that EGFR had no influence on prognosis. It is possible that altered EGFR expression may be associated with local recurrence, which is more frequently life threatening at other sites. HER-2 was negative in all samples and may play little part in CSCC progression as found in squamous cell carcinomas from other sites. High HER-4 expression in lymph node metastases was associated with poor prognosis suggesting a role in progression of cSCC of the trunk and extremities. It is possible that altered HER-4 expression occurs late and is present only in metastases. The altered coexpression of the HER family may play a role (i.e., EGFR/HER-4, HER-3/HER-4, and EGFR/HER-3/HER-4), but the small number of cases in this study meant this could not be analyzed.
Lymphatic Vessel Markers: Podoplanin
Podoplanin, originally detected on the surface of podocytes, belongs to the family of type-1 transmembrane sialomucin-like glycoproteins. Although specific for lymphatic vascular (LV) endothelium, podoplanin is expressed in a wide variety of normal and tumor cells. Podoplanin plays an important role in preventing cellular adhesion and is involved in the regulation of the shape of podocyte foot processes and in the maintenance of glomerular permeability. Moreover, podoplanin is involved in LV formation and does not influence formation of blood vessels.
The expression of podoplanin is induced by the homeobox gene Prox-1, and a specific endogenous receptor was identified on platelets. Immuno histochemical detection of podoplanin/D2-40 in LECs was used in many studies to evaluate the LV microvascular density (LVMD) in peritumoral and tumoral areas and to correlate LVMD with lymph node status and prognosis. Podoplanin significantly increases the detection of lymphovascular invasion in different types of malignant tumors. Podoplanin expression was found in tumor cells of various types of cancer, such as vascular tumors, malignant mesothelioma, tumors of the central nervous system (CNS), germ cell tumors, and squamous cell carcinomas. This expression in tumor cells is useful for pathological diagnosis, and podoplanin seems to be expressed by aggressive tumors, with higher invasive and metastatic potential [72]. Recently, it was demonstrated that podoplanin expression was not associated with the presence of lymph node metastasis, but was a prognosticator of reduced survival indicating a locally aggressive tumor, with survival impact in cSCC. Altered expression of podoplanin is associated with mesothelioma, squamous cell carcinoma of oral mucosa, and germ cell tumors, suggesting that podoplanin may influence invasive and proliferative activity. As cSCC metastases occur preferentially via lymphatic vessels, podoplanin expression may be associated with disease progression. Hyperexpression has been related to undifferentiated skin tumors, but its impact on prognosis and metastasis has not been established. Podoplanin can be suggested as a possible target for the development of novel therapies, and its expression has to be studied in other settings to completely understand its role in cSCC development and progression.
Progression of cSCC
Progression of cSCC is substantially due to activation of proto-oncogenes such as ras and inactivation of tumor suppressor genes such as PTCH or INK4a-ARF. Moreover, epigenetic deregulations have also been described as an important determinant in the progression of cSCC.
Activation of ras Oncogenes
The members of ras family, H-ras, K-ras, and N-ras, encode 21 kDa guanosine triphosphate (GTP)-binding proteins placed on the inner surface of the cell membrane [73]. They are functional in the transduction of signals to the nucleus in order to activate proliferation machinery, when growth factors are activated through intrinsic GTPase. Different human cancers show ras mutations in codons 12, 13, and 61 [74] with consequent continuous activation of ras-mediated signal transduction. Also human skin cancers contain mutations in all three members of the ras family [75].
Activated c-H-ras oncogenes derived from UV-exposed cSCC are tumorigenic for NIH 3T3 cells [76]. Therefore, the transfected NIH 3T3 cells induce tumors at the subcutaneous site of injection and spontaneous lung metastases in nude mice [77]. Also ras family mutations rely at pyrimidine-rich sequences, suggesting a direct role of UV irradiation [78]. Besides point mutations, deregulation of ras genes could be due to amplification and rearrangement of specific gene sequences [79, 80].
Mutations in PATCH and INK4a/ARF Tumor Suppressor Genes
Mutations in INK4a/ARF tumor suppressor gene have been described in a subset of cSCCs. This locus encodes for two independent growth inhibitors: the cyclin-dependent kinase (CDK) inhibitor p16INK4a and the p53 activator p14ARF (mouse p19Arf). p16INK4a inactivates CDK4/6, favoring sequestration of the transcription factor E2F1 by the Rb tumor suppressor protein [81]. p14ARF binds human double-minute protein 2 (HDM2, mouse Mdm2), thereby blocking HDM2-induced translational silencing and degradation of p53 [82]. Also in these cases gene alterations were induced by UV. Recently, in a series of cSCC, all the alterations of the INK4a/ARF locus have been analyzed. Point mutations were less common (10 %) and loss of heterozygosity (32.5 %) and promoter hypermethylation of p16INK4a and p14ARF (36 %) more common [83].
Epigenetic Alterations
Epigenetic alterations described in cancer progression include DNA methylation and histone modifications, the latter consisting in methylation, acetylation, phosphorylation, ubiquitination, and sumoylation, chromatin remodelling and different microRNA profile [84–86]. Gene specific methylation responsible of transcriptional control is described in cSCC development and progression [84–86]. Promoter methylation of FOXE1, a transcriptional factor involved in regulation of many organ development and located on chromosome 9q22, a region frequently lost in cSCC, is highly methylated in cSCC [87]. microRNAs (MiRNAs) appear to play an important role in the epigenetic regulation of cSCC at posttranscriptional level [88]. In fact, a recent study has demonstrated that a distinct microRNA profile is specifically induced by UV radiation [89]. Finally, mitochondrial DNA mutations, affecting displacement loop (D-loop) and other regions, are described in cSCC [90–92].
Human Papillomavirus (HPV) and cSCC
In cSCC carcinogenesis, also HPV infection appears to play an important role significantly reducing p53 expression, particularly in immune-suppressed patients [93]. In details, higher viral load has been recorded in 80 % of cSCC of immunosuppressed organ transplant patients [20], while in immune-competent patients, the frequency of HPV DNA in cSCC varies from only 27 to 70 % depending on detection techniques [23, 29]. On this light, among more than 100 HPV identified subtypes, only the small subgroup of high-risk mucotropic HPV (HPV types 16, 18, 31, 33, 35, and 58) has been demonstrated as responsible of cervical cancer development. In this context E6 gene favors p53 proteosomal degradation with subsequent abolishment of cell cycle arrest or apoptosis.
HPV DNA is often identified in cSCC including more than 40 not high-risk subtypes [94, 95]. The pathogenesis of HPV-induced cSCC is probably different from that observed in cervical cSCC. In fact, low copy of HPV DNA has been found in cSCC, suggesting a possible role for tumor initiation and progression. In details, three mechanisms have been proposed for HPV-induced carcinogenesis: (1) UV radiation-induced immunosuppression that favors HPV infection [96, 97], (2) E6/E7 oncoprotein-related changes in p53 and Rb tumor suppressor gene, and (3) integration of HPV DNA disrupting genomic stability [96]. However, the weak transforming activity of cutaneous HPV in vitro suggests that antiapoptotic activity of E6 protein in cSCC could become relevant only in conjunction with other factors, such as by sunlight (UV) exposure, immunosuppression, and proliferation of epithelium (psoriasis) [95] (Fig. 3.2).
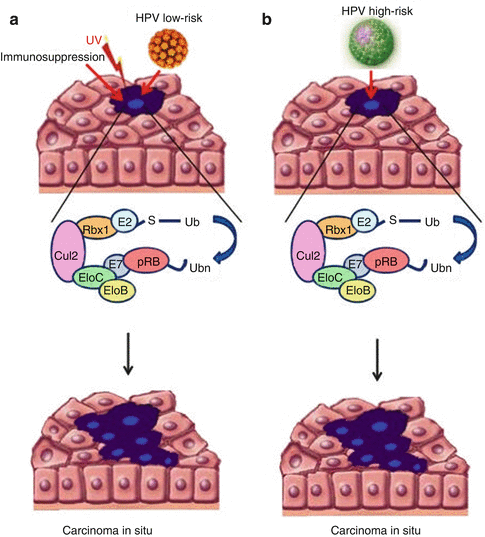
Fig. 3.2
Mechanisms of HPV-induced cSCC: Low-risk HPV (a) needs cofactors to induce cSCC, differently from high-risk HPV (b)
These hypotheses are supported by the evidence that HPV-38, detected in 50 % of skin carcinomas, in 43 % of AK, and in 10 % of nonneoplastic keratinocytes, is responsible of longevity/immortalization of human keratinocytes in vitro [97–99].
Clinicopathological data of HPV-related cSCC show close association with poorly differentiated cancers, lymph nodes metastasis, and late-stage disease, which are related to poor prognosis tumors. HPV status is also characterized by p16 overexpression and low frequency of p53 mutations [100].
Histology
cSCC includes a series of neoplastic disorders with substantial biological differences. They could be grouped in two categories: sun exposure-related tumors, such as preinvasive lesions (actinic or solar keratoses (AKs), cSCC in situ(Bowen’s disease)) and invasive lesions (invasive cSCC (cSCCI), clear-cell cSCC, spindle cell (sarcomatoid) cSCC, and cSCC with single-cell infiltrates) that derive from preinvasive lesions, and non-sun exposure-related lesions, such as de novo cSCC, lymphoepithelioma-like carcinoma of the skin (LELCS), and verrucous carcinoma (VC), not related to solar exposure.
Actinic Keratosis (AK)
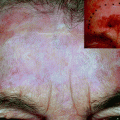
It is commonly considered a cSCC precursor lesion. It is related to solar exposure; therefore, it develops in sun-exposed skin areas, including face, neck, dorsal hands, and forearms, upper chest, back, and scalp [4, 5, 15, 101]. Due to its relation to exposure to sun, it generally affects middle-aged or older individuals. It can occur in younger individuals, but always in relation to longtime sun exposure or concomitant sensibility to UV damage, such as immunosuppression [15, 24]. AK spontaneously regresses and evolves to invasive cSCC in only 5–10 % of cases [102]. Different AK variants have been described including hypertrophic, atrophic, acantholytic, pigmented, proliferative, and bowenoid subtypes. Hypertrophic and proliferative variants are associated to a more aggressive biologic behavior [4, 5, 103, 104].
It is characterized by aggregates of atypical, pleomorphic keratinocytes that show nuclear atypia, dyskeratosis, and loss of polarity, extending variously through the epithelium till to cover all the thickness in the bowenoid variant, similar to Bowen’s disease [5, 105, 106] (Fig. 3.3a). The profile of dermo-epidermal junction in AKs shows small round epithelial buds protruding slightly into the upper papillary dermis. Solar elastosis in the dermis is always observed.
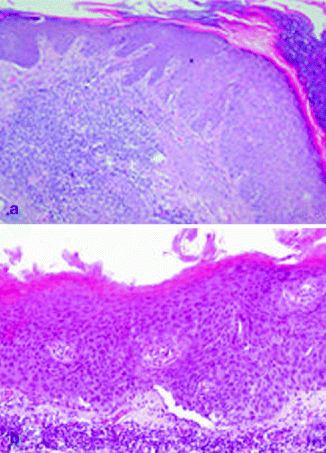
Fig. 3.3
(a) Actinic keratosis, H&E 10×; (b) Bowen’s disease, H&E 20×
Squamous Cell Carcinoma In Situ (cSCCS)/Bowen’s Disease
The two definitions are synonymous. Most of them occur in sun-exposed skin areas, such as the head, neck, and hands [107]. Moreover, mucosal surfaces and nail could be frequently involved [108]. Bowen’s disease in genital areas is also called erythroplasia of Queyrat. Clinical presentation is generally represented by erythematous, scaly patch or plaque [108]. Only 3–5 % of cases progress towards invasive cSCC, and this rate is higher for erythroplasia of Queyrat [109]. The invasive cSCC developed from Bowen’s disease seems to be more aggressive, since 20 % of these cases develop metastatic disease [110]. Histopatho logically, the epidermis is characterized by atypical cells with loss of polarity, high mitotic activity, pleomorphism, and greatly enlarged nuclei through the entire thickness (Fig. 3.3b). Multinucleated cells could be also observed [107]. Atypical cells may involve adjacent follicular epithelium and adnexal structures [5].
Invasive Squamous Cell Carcinoma (cSCCI)
Approximately 97 % of cSCC are associated to AK as a consequence of their strict biological link. Also histological features of cSCC are similar to its precursor but characterized by infiltrative pattern of membrane basement into dermis [3, 111] (Fig. 3.4). Infiltrative dermal component is represented by neoplastic cell nests, often associated to inflammatory infiltrate. cSCCI differentiation is defined by three broad histologic grades, based essentially on nuclear atypia and keratinization (Fig. 3.5a–c). Approximately 0.5 % of metastases has been recorded for well-differentiated tumors, containing cells with slightly enlarged, hyperchromatic nuclei with abundant amounts of cytoplasm [15]. On the contrary, poorly differentiated tumors, containing highly pleomorphic cells with scanty keratin formation, are characterized by much more aggressive clinical behavior [16]. Intermediate differentiated cSCCI share features of both well-differentiated and poorly differentiated tumors.
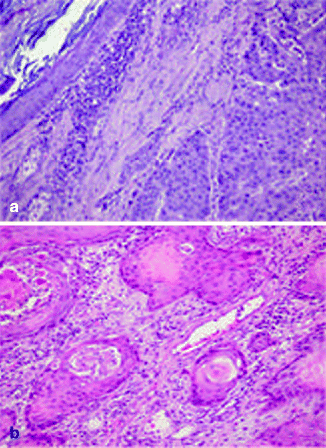
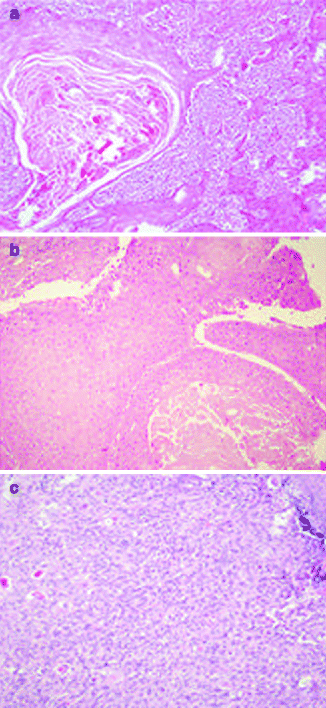
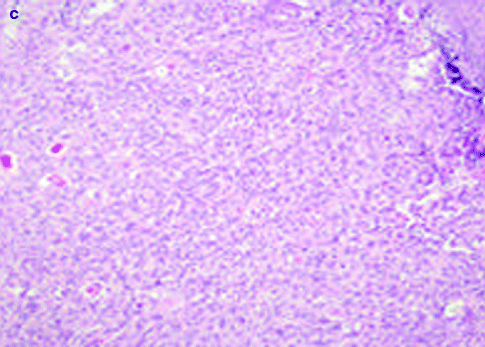
Fig. 3.4
(a) cSCC in photo-exposed area with the concomitant presence of solar elastosis, H&E 20×; (b) SCC in not photo-exposed area, H&E 20×
Fig. 3.5
Well-differentiated (a), moderately differentiated (b), and undifferentiated (c) cSCC, H&E 20×
Clear-Cell cSCC
This rare variant shows neoplastic cells with clear cytoplasm, because of hydropic degeneration of neoplastic cells, and the accumulation of intracellular fluid [112]. Clinically, the lesions as nodules or ulcerated masses are typically present in sun-exposed surfaces. Histopathologically, three different types have been recognized: (1) keratinizing (type I), (2) nonkeratinizing (type II), and (3) pleomorphic (type III). All three types are characterized by clear cytoplasm without glycogen or mucin, but only trace of lipid, suggesting that clear-cell changes are degenerative [112].
Type I lesions are characterized as sheets or islands of tumor cells with clear, empty-appearing cytoplasm and foci of keratinization. Type II lesions are dermal tumor without keratinizing foci, while type III lesions arise from the epidermis, widely ulcerated, with foci of squamous differentiation, areas of acantholysis, and the presence of dyskeratosis (Fig. 3.6a). Its rarity makes it difficult to distinguish histologic markers of aggressiveness.
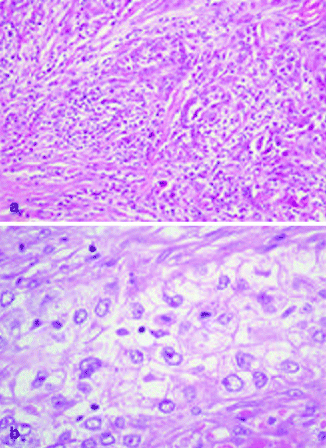
Fig. 3.6
(a) Sarcomatoid cSCC, H&E 40×; (b) Clear-cell SCC, H&E 40×
Spindle Cell (Sarcomatoid) cSCC
Spindle cell cSCC is a very rare variant of squamous cell carcinoma [113]. It has been observed in sun-exposed areas, but it can develop also in patients with prior exposure to radiation. In the latter case, this variant is considered de novo cSCC and appears to be more aggressive than conventional cSCC [113]. Clinically it is characterized by either bleeding or ulceration. Microscopically spindle cells cSCC is composed by tumoral spindle cells arranged in whorled pattern (Fig. 3.6b). It can be also associated to conventional cSCC and AK [114]. Neoplastic cells can contain also highly pleomorphic giant cells and heterologous elements. The tumor is generally deeply infiltrating, sometimes with the involvement of subcutis, fascia, muscle, and even occasionally bone. High mitotic index is generally present. Immunohistochemical staining is needed, in the absence of conventional cSCC, in order to distinguish it from other spindle cell tumors, such as atypical fibroxanthoma, spindle cell melanoma, or spindle cell sarcoma. Spindle cell cSCC stains, at least focally, high molecular weight cytokeratin and p63 [115].
cSCC with Single-Cell Infiltrates
This is a rare entity, occurring on the face and neck of older individuals. It seems more aggressive than conventional cSCC because it is frequently misdiagnosed [116]. Microscopically, it is formed by loosely arranged neoplastic cells in the dermis, without apparent connection to epidermis or adnexal structures. The diagnosis is very difficult, above all when neoplastic cells are obscured by inflammatory infiltrates. Immunohistochemical staining of neoplastic cells for high molecular weight cytokeratin and p63 allows the recognition of these cells [116].
De Novo cSCC
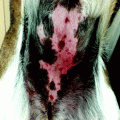
De novo cSCC generally arises on skin surface previously injured or diseased, particularly long-standing ulcers, burn scars, or osteomyelitis, and also chronic inflammatory conditions such as discoid lupus erythematosus and dystrophic epidermolysis bullosa [117–120]. Marjolin’s ulcer refers to cSCC developed in burn scars, first described by Marjolin in 1827 [121]. De novo cSCCs are commonly found on the lower extremities, where burn scars are more prevalent. The time of incidence from the trauma is around 20 to 40 years later [122]. Clinically it is presented as exophytic growths or indurated ulcers. Microscopically, de novo cSCCs are similar to conventional cSCC, with the lack of solar dermis elastosis (Fig. 3.4). It is associated to either ulcers or scars. The epidermis is often atrophic or ulcerated. The incidence of regional lymph node metastasis, particularly for lower extremity lesions, is higher if compared with other cSCC subtypes, reaching as high as 54 % [122]. Its prognosis is very poor, with a 5-year survival rate of only 52–75 % [5].
Verrucous Carcinomas
Verrucous carcinoma (VC) is a relative indolent noninfiltrating tumor, associated with both low-risk (types 6 and 11) and high-risk (types 16 and 18) types of HPV [23]. Four clinicopathological categories based on the involved body skin area are described: oro-aero-digestive VC, ano-urogenital VC, palmoplantar VC (also known as epithelioma cuniculatum), and VC found on other sites [123]. Despite this subclassification, all these lesions share similar histological features.
As cutaneous VC concerns, the most frequent category is palmoplantar VC, found on the soles of elderly Caucasian males, but it may also be seen on the toes, the heel, or the dorsum of the foot [124]. They are frequently misdiagnosed as plantar warts, but they finally emerge as bulky exophytic masses. Low-risk HPV types is commonly found in thesetumors [123].
VC lesions found on other cutaneous sites are rarely describes. They are clinically similar to palmoplantar VC, but rarely associated to HPV infections [123].
Microscopically, VC are well-differentiated tumors, with a characteristic endo-exophytic growth pattern, normal stratification of squamous cells, marked acanthosis and papillomatosis, hyperkeratosis with parakeratosis, and a prominent granular layer. Minimal atypia can be also present (Fig. 3.7). Tumoral projections through the dermis descend deeply, and they seem to be responsible of compressive destruction [123].
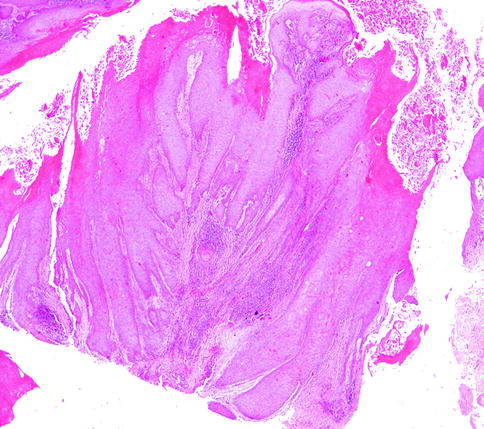
Fig. 3.7
Verrucous carcinoma, H&E 5×
The distinction between conventional cSCC and VC is very relevant, conventional cSCC being more aggressive. Immunohistochemical staining of bcl-2, Ki-67, and p53 of the lower third of the epidermis in VC and the entire epidermis in cSCC is very useful for the differential diagnosis [125].
Lymphoepithelioma-Like Carcinoma of the Skin
This rare entity occurs in the head and neck of elderly patients. Clinically it is a slowly growing dermal nodule. Histologically it is characterized by syncytial sheets of tumor cells in the mid to deep dermis in an inflammatory background [126]. Only electronic microscopy has demonstrated its squamous origin, due to the presence of desmosomes and tonofilaments [127]. Also in these cases, the diagnosis of neoplastic cells can be difficult, because they are obscured by inflammatory elements. Also in this case, the immunohistochemical staining of neoplastic cells for epithelial markers, such as cytokeratin and EMA, can facilitate the recognition of neoplastic cells [128].
Therapeutic Approaches Based upon the Targeting of Biological Markers Expressed in cSCC
Treatment of Nonadvanced cSCC
For low-risk, local lesions, the usual treatment is surgical excision, electrodessication and curettage, or cryosurgery. Destructive treatment methods leave no tissue to analyze for marginal control. Nevertheless, using these methods, the 5-year control rate in patients with low-risk primary lesions can be as high as 96 %.
For higher-risk tumors, the primary treatment is surgical excision. The key factor in determining the cure rate is the ability to achieve negative surgical margins. Surgery may be conventional or microscopically controlled, the latter procedure referred to as “Mohs’ surgery,” after its originator. In this procedure, the targeted lesion is excised and the circumferential margins are assessed microscopically for residual tumor. Margins remaining involved undergo repeated excisions, followed by histological assessment, until negative margins are obtained. Mohs’ surgery yields local control rates of 92–100 %, versus 38–87 % for standard surgical excision. Mohs’ surgery cure rates decrease as tumor grade increases, with a 45.2 % cure rate for grade 4 cSCC.
Treatment of Advanced cSCC
Chemotherapy
A number of systemic therapies have been used to treat advanced cutaneous SCC, including cytotoxic chemotherapy (cisplatin, 5-fluorouracil [5-FU], bleomycin, and doxorubicin), 13- cis-retinoic acid (13cRA), and immunotherapy (interferon-α 2a [IFN-α]).
A nonrigorous randomized trial comparing bleomycin with other cytotoxic agents (cyclophosphamide, vincristine, methotrexate, and procarbazine) as treatment for 70 patients with SCC, only six of whom had cSCC, showed no statistically significant difference between the two treatment groups [129]. Sadek et al. [130] reported on 14 patients (13 evaluable) from a prospective observational study of patients with advanced cutaneous SCC treated with cisplatin, 5-FU, and bleomycin for 1–4 months. That study resulted in 4 of 13 patients with a complete response (CR) and 7 of 13 with a partial response (PR) [131]. After 1 year, 6 of 13 patients had died from their disease and 6 of 13 had no evidence of disease.
Two case series reported patients achieving a CR with a PR that was achieved in three of seven patients. The median duration of CR was 1 year. Fujisawa et al. [132] reported on two patients achieving a CR after one or two cycles of cisplatin and 5-FU. This therapy seemed to be effective in terms of local control, but was ineffective in preventing metastases.
Guthrie et al. [133] treated three SCC patients with cisplatin and doxorubicin. One patient had a CR for 17 months, one had a PR for 3 months, and one had stable disease (SD). The authors suggested that the combination of cisplatin and doxorubicin had activity in cSCC. A phase II trial by the same group used this regimen in 12 cSCC patients [134]. Interpretation of that trial’s results was hampered by the low numbers of enrolled patients and their heterogeneity. Seven patients were treated with chemotherapy alone: two achieved a CR for 4–12 months, two achieved a PR for 3–6 months, and three had no response. Five patients received neoadjuvant chemotherapy followed by surgery or definitive radiation therapy, with two CRs and one PR after induction chemotherapy. The authors concluded that this combination had activity in advanced cutaneous SCC patients and that multimodality therapy was to be preferred over chemotherapy alone.
Wollina et al. [135] used oral capecitabine and s.c. IFN-α in four patients with advanced cutaneous SCC. They reported two patients with CRs and two patients with PRs using this regimen.
Oral 5-FU was administered to 14 patients with advanced cSCC as a single agent [136]. Two patients experienced a PR and seven had SD of varying duration. This report, however, supports the use of fluoropyrimidine-based therapy in advanced cSCC patients, because there may be palliative benefit even with the use of single-agent therapy.
Retinoids and IFN-α in Cutaneous SCC
Retinoids modulate cell differentiation and proliferation; in vitro, some cytokines can act synergistically with retinoids to inhibit cell proliferation and increase apoptosis. Shin et al. [137] assessed the effects of IFN-α, 13cis retinoic acid (13cRA), and cisplatin used to treat unresectable SCC of the skin in a prospective phase II trial. Six of 35 (17 %) patients experienced a CR and 6 of 35 (17 %) had a PR. The response rate of patients with locoregional disease was higher (67 %) than that of patients with metastatic disease (17 %; P = .007). They concluded that this combination was useful in treating locally advanced disease, but less so in metastatic disease.
Lippman et al. used 13cRA in combination with IFN-α in a prospective phase II trial enrolling 32 patients with inoperable cSCC. They observed a response in 19 (68 %) of the 28 evaluable patients (7 CRs, 12 PRs). The median duration of response was 5 months. Response rates varied with the extent of disease: 93 % (13 of 14) responded among patients with advanced local disease, 67 % (4 of 6) responded among patients with regional disease, and 25 % (2 of 8) responded among those with distant metastases. This combination appeared to be effective in advanced SCC patients, albeit with greater efficacy in less advanced disease.
Although not focused on treatment of unresectable disease, Brewster and coworkers reported on a phase III trial testing whether adjuvant therapy with 13cRA and IFN-α was effective in preventing recurrences and increasing time to recurrence. Their adjuvant study enrolled 66 patients with “aggressive” SCC. The 66 patients were randomly assigned, following initial surgery, to receive either a combination of 13cRA and IFN-α for 6 months or no systemic adjuvant therapy. Adjuvant radiotherapy was added to the initial treatment plan for tumors with perineural invasion, more than two positive nodes, extracapsular nodal disease, or microscopically positive margins. With a median follow-up of 21.5 months, this systemic treatment did not improve time to recurrence or prevent secondary tumors.
Anti-EGFR Therapy in Cutaneous SCC
Cetuximab, a humanized monoclonal antibody, inhibits EGFR by blocking the extracellular domain of EGFR. This prevents the receptor’s ligand from binding and consequent dimerization. One phase II study and two case reports have described its effects in cSCC. In a phase II trial enrolling 36 naïve patients with unresectable or metastatic cutaneous SCC that expressed EGFR [138], 31 patients of 36 enrolled were evaluable for tumor response. The disease control rate (DCR) after 6 weeks of treatment was 69 %, and the overall response rate was 11 %. The mean progression-free survival (PFS) and overall survival (OS) times were 121 days and 246 days, respectively. Among the 31 evaluable patients, development of an acneiform rash did not predict response to treatment, but did predict the mean PFS and OS times. Such drug rashes have been associated with better outcome in other diseases treated with cetuximab, such as SCC of the head and neck.
Randomized trials of cetuximab in metastatic colorectal cancer (mCRC) patients have confirmed the importance of mutational status in the signal transduction apparatus downstream from EGFR, including KRAS and BRAF, and in an immune marker, the Fc antibody receptor [139, 140]. In a subset of 28 patients studied by Maubec and coworkers, mutational status was assessed in exon 2 (n = 28) and exon 3 (n = 25) of KRAS and exon 15 (n = 23) of BRAF kinase. All were found to be wild type. Cutaneous SCC patients possessing the FcγIIa-131 H/H or FcγIIIa-158 V/V variants, associated with better outcome in mCRC patients, had a PFS interval similar to the wild-type 131R and 158 F carriers in the Maubec et al. study [138].
Two case reports of cSCC patients treated with cetuximab have also been published, both achieving CRs [141, 142]. More recently, eight cases of locoregionally advanced or unresectable cSCC treated with either cetuximab alone or cetuximab and radiotherapy and followed up for 23 months were presented. Two patients were treated with cetuximab as single-agent therapy, and four were treated with cetuximab and concomitant radiotherapy. Either cetuximab alone or the combination of cetuximab and radiotherapy can be effective in cSCC, with 6/8 responses, including three CRs and a median OS of 22.5 months. Moreover, the use of cetuximab and radiotherapy according to previously reported modalities seems to be more efficient than single-agent use, with 3 CRs observed in this series. It is noteworthy that most of this study had a very poor prognosis, having received heavy previous surgery and/or radiotherapy for several previous relapses.
Gefitinib inhibits binding to the ATP-binding site of EGFR, rendering it unable to autophosphorylate and activate the receptor. Glisson et al. [143] used gefitinib in a prospective phase II trial, enrolling 18 patients with advanced or recurrent cutaneous SCC. Gefitinib had already been reported to have an 11 % response rate and 53 % control rate in head and neck SCC patients [144]. Four of the 15 evaluable cSCC patients had SD after 4 weeks of treatment.
Erlotinib, much like gefitinib, competitively binds to the ATP-binding site of EGFR. It has been approved for use in non-small cell lung cancer patients who have failed to respond to chemotherapy, and in advanced pancreatic cancer patients, combined with gemcitabine. Read et al. [145] reported results from two patients with unresectable cSCC. One patient achieved a CR after 1 month of treatment and the other achieved a PR after 3 months of treatment. The patient that achieved a CR had a recurrence when the therapy was discontinued.
cSCC Secondary to Raf Kinase Inhibitors
The T → A transversion at position 1799 of BRAF (BRAF V600E) is present in approximately 50 % of patients with metastatic melanoma [146, 147]. Recently, it was also demonstrated that this mutation can be present in about 25 % of the patients affected by hepatocellular carcinoma in which medical treatment with Raf inhibitor have received approval by US and European agencies. BRAF V600E induces constitutive signaling through the MAPK pathway, stimulating cancer cell proliferation and survival [147]. The clinical development of inhibitors of oncogenic BRAF, termed type I BRAF inhibitors, which block the active conformation of the BRAF kinase, has led to a high rate of objective tumor responses and improvement in overall survival, as compared with standard chemotherapy in melanoma [148, 149]. However, nonmelanoma skin cancers – cSCC and keratoacanthomas (KA) – have developed in approximately 15–30 % of patients treated with type I BRAF inhibitors such as vemurafenib and dabrafenib (GSK-2118436) [150, 151].
KA and cSCC most frequently develop within 8–12 weeks of beginning therapy. Similar treatment-related skin neoplasms have been described with the structurally unrelated multikinase inhibitor sorafenib [152, 153]. Sorafenib has been reported to have pan-RAF inhibitory properties [154], although the overall cellular potency of this compound against RAF proteins is much less pronounced when compared with selective inhibitors [155]. Perhaps not surprisingly, sorafenib-induced skin tumors occur much less frequently and are more delayed in onset [152, 153]. Together, these observations suggest that RAF inhibition may play a direct role in the development of skin tumors.
Recently, four international centers contributed 237 KA or cSCC tumor samples from patients receiving an RAF inhibitor (either vemurafenib or sorafenib; n = 19) or immunosuppression therapy (n = 53) or tumors that developed spontaneously (n = 165). Each sample was profiled for 396 known somatic mutations across 33 cancer-related genes.
Mutations were detected in 16 % of tumors (38 of 237), with five tumors harboring two mutations. Mutations in TP53, CDKN2A, HRAS, KRAS, and PIK3CA were previously described in squamous cell tumors. Mutations in MYC, FGFR3, and VHL were identified for the first time. A higher frequency of activating RAS mutations was found in tumors from patients treated with an RAF inhibitor versus populations treated with a non-RAF inhibitor (21.1 % v 3.2 %; P = .01), although overall mutation rates between treatment groups were similar (RAF inhibitor, 21.1 %; immunosuppression, 18.9 %; and spontaneous, 17.6 %; P = not significant). Tumor histology (KA v cSCC), tumor site (head and neck v other), patient age (>70 v <70 years), and sex had no significant impact on mutation rate or type.
In another study, a molecular analysis to identify oncogenic mutations (HRAS, KRAS, NRAS, CDKN2A, and TP53) in the lesions from patients treated with the BRAF inhibitor vemurafenib was performed.
Among 21 tumor samples, 13 had RAS mutations (12 in HRAS). In a validation set of 14 samples, 8 had RAS mutations (4 in HRAS). Thus, 60 % (21 of 35) of the specimens harbored RAS mutations, the most prevalent being the activating HRAS Q61L. Increased proliferation of HRAS Q61L-mutant cell lines exposed to vemurafenib was associated with MAPK-pathway signaling and activation of ERK-mediated transcription. In a mouse model of HRAS Q61L-mediated skin carcinogenesis, the vemurafenib analogue PLX4720 was not an initiator or a promoter of carcinogenesis but accelerated growth of the lesions harboring HRAS mutations, and this growth was blocked by concomitant treatment with a MEK inhibitor.
The paradoxical hyperactivation of MAPK by RAF inhibitors predicts that upstream oncogenic events, either activating mutations in RAS or mutations or amplifications in receptor tyrosine kinases that strongly elevate levels of the RAS–GTP complex in the absence of a BRAF V600E mutation, would potentiate signaling through the MAPK pathway [156, 157]. Some preclinical experimental models have demonstrated that signaling occurred preferentially through CRAF, with RAF inhibitors thought to induce a conformational change in CRAF/BRAF heterodimers or BRAF homodimers that resulted in pathway hyperactivity [158, 159]. The functional studies of the previous report showing HRAS-primed activation of the MAPK pathway in models of SCC treated with BRAF inhibitors provide evidence that the toxicity related to BRAF inhibition may arise from paradoxical MAPK-pathway activation in humans. Recent studies have shown that vemurafenib resistance can be mediated by receptor tyrosine kinases such as the platelet-derived growth factor and insulin-like growth factor 1 receptors [160, 161]. Preexisting amplification of the EGFR gene in the A431 cell-line model also resulted in paradoxical MAPK-pathway signaling in functional assays, although at a lower level than that driven by oncogenic HRAS. These data from in vitro models suggest that similar mutations or amplifications of receptor tyrosine kinases may account for the development of cSCC and KA in the 40 % of samples in which no RAS mutations were found.
The timing of the appearance of these lesions after vemurafenib treatment is decidedly different from that of secondary cancers associated with cytotoxic chemotherapy. In the case of vemurafenib, the lesions tend to appear within the first few weeks after the start of therapy, whereas cancers that are due to the genotoxic effects of chemotherapy develop years after exposure. The specificity of vemurafenib for a limited number of kinases [162, 163], along with the finding that RAS mutations occur frequently in lesions arising preferentially in sun-damaged skin, suggests that vemurafenib may not have direct carcinogenic effects but instead may potentiate preexisting initiating oncogenic events.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


