Summary of Key Points
- •
Lung cancers commonly have structural chromosome aberrations and aneuploidy, with many of them associated with carcinogenesis.
- •
Gene amplification is a common mechanism of oncogenic activation in nonsmall cell lung cancer (NSCLC) involving genes such as MYC , EGFR , ERBB 2, MET , PIK3CA, and FGFR1. Gene amplification is also associated with resistance to drugs, for instance, to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors when the T790M EGFR allele or the MET gene is amplified.
- •
More recently, genes such as ALK , ROS1 , RET , and NTRK1 were found to be activated in NSCLC by fusions with gene partners subjected to constitutive transcription or carrying specific domains inducing phosphorylation.
- •
Novel therapeutics have been developed to target those specific molecular drivers, and several drugs have succeeded in substantially improving survival and quality of life of patients carrying such molecular changes.
- •
NSCLC tumor profiling is achieved by numerous technologies focusing on different levels such as DNA (e.g., sequencing, fluorescence in situ hybridization [FISH]), RNA (e.g., reverse transcription-polymerase chain reaction), and protein (e.g., immunohistochemistry [IHC]). Technologies using in situ (FISH, IHC) and extraction (PCR based, sequencing) platforms have distinct advantages and limitations.
- •
Single tests and test panels are available, with the latter being most effective due to the low incidence of the rearrangements in the overall NSCLC population, the lower cost per gene tested, and the scarcity of tumor tissue in patients with advanced stage disease. Molecular drivers have been detected more commonly in lung adenocarcinomas than in squamous cell carcinomas (SCCs) but a strong effort is ongoing to better define potential therapeutic targets in SCC. Very little is known regarding markers for therapy in small cell lung cancer.
Lung cancer is a group of diseases displaying a high level of genomic instability and complex molecular changes. This chapter reviews the impact of molecular events detectable by cytogenetic techniques, such as instability, at the DNA and chromosome levels in lung cancer patients. In addition, the chapter also examines two of the major molecular mechanisms leading to the nonviral activation of oncogenes: gene amplification and gene fusion. Overexpression of proteins in conditions in which they are normally absent is commonly driven by an increase in gene copy number or the release of a specific gene from the ligand binding control, thus having its active domain under the control of the promoter or of the active domain of a constitutively activated gene. Amplification of genes in lung cancer was discovered in the mid-1980s for v-myc avian myelocytomatosis viral oncogene homolog ( MYC ) and Kirsten rat sarcoma viral oncogene homolog ( KRAS ). Conversely, fusion proteins are a much newer phenomenon in lung cancer. Despite being common and well-known in leukemia and lymphoma as causal factors and targets for therapy, gene fusions were not described in lung cancer research before the start of the 21st century. However, with the progress of genomic technology, the number of activated gene fusions discovered mainly in NSCLC has increased rapidly, as will be detailed here.
Genetic Instability in Lung Cancer
Accumulation of multiple genetic abnormalities is known to be associated with lung cancer initiation and progression. Genetic instability, which may cause these abnormalities, is a general term that refers to both chromosomal instability (CIN) and microsatellite instability (MSI). Instability involving whole or partial regions of chromosomes (CIN) includes deletion, duplication, insertion, and translocation. CIN may induce loss of heterozygosity (LOH) of tumor suppressor genes or DNA repair genes when deletions occur, and amplification of oncogenes by multiple duplications of focal chromosome regions. Consequently, compelling evidence has supported the role of CIN in the pathogenesis of lung cancer. In addition to alterations at the chromosomal level, instability at the nucleotide level, frequently referred to as MSI, is usually connected to mismatch repair (MMR) defects. MSI may cause missense mutations that facilitate the inactivation of tumor suppressor genes, such as p53 , which may contribute to the development and progression of lung cancer. Both phenomena—CIN and MSI—obviously contribute to the phenotype instability and versatility of cancer cells. Therefore an understanding of the molecular mechanisms leading to genetic instability holds promise for the development of novel therapeutic strategies in lung cancer.
Microsatellite Instability
Microsatellites, also known as simple sequence repeats, are tandem repeats of short (fewer than 10 bp) DNA sequences, which are useful markers for genetic mapping and LOH of defined chromosomal loci. The most common microsatellite in humans is a dinucleotide repeat of CA, which occurs tens of thousands of times across the genome. Although the length of these microsatellites is highly variable from person to person, each individual has microsatellites of a set length. MSI, a hallmark of genetic instability, generally occurs because of abnormalities of the MMR genes, such as hMSH2 and hMLH1 , impairing the correction of errors that spontaneously occur during DNA replication. The loss of MMR function renders tumor cells susceptible to the acquisition of somatic mutations throughout the genome, and microsatellites are particularly susceptible to mutations in the absence of MMR. MSI was initially identified in colorectal cancer and was immediately clinically significant because of its association with hereditary nonpolyposis colon cancer (HNPCC). In HNPCC, the MSI of MMR genes due to germline alterations is an essential molecular basis of its development. By contrast, in lung cancer, CIN plays a more important role in carcinogenesis, as homozygous and heterozygous deletions of certain chromosomal loci or amplification of oncogenes frequently occur, as will be described.
There are conflicting data on the relevance of MSI in lung cancer. The frequency of MSI has been reported to range from 0% to 69% in NSCLC and from 0% to 76% in small cell lung cancer (SCLC). Interestingly, several studies of NSCLC have demonstrated a higher frequency of MSI in tetranucleotide-repeating regions than in traditional mononucleotide-repeating or dinucleotide-repeating regions, and the term “elevated microsatellite alterations at selected tetranucleotide” (EMAST) has been proposed to designate the phenomenon. Furthermore, EMAST was reported to be associated with SCC with lymph node metastasis. The molecular mechanisms leading to EMAST, distinct from traditional MSI, were not associated with defects in MMR, but it was suggested that p53 alterations may be involved.
Aneuploidy and CIN
Most cancer cells possess an abnormal number of chromosomes, often in the triploid or tetraploid range. In addition to the altered number of chromosomes, cancer cells commonly have structural chromosome aberrations, such as inversions, deletions, duplications, and translocations. Aneuploidy, defined as numerical and structural abnormalities of chromosomes, commonly results from CIN. Aneuploidy and CIN can mediate the evolution of cancer cell populations under selection pressure and are associated with poor prognosis and distinctive histopathologic features in many tumors. CIN plays an important role in lung carcinogenesis by accelerating homozygous and heterozygous deletions of tumor suppressor genes and effectively amplifying oncogenes. Therefore a better understanding of the causes and effects of aneuploidy and CIN may lead to new therapeutic venues for solid malignancies, including lung cancer.
Early studies have shown that lung cancer frequently exhibits marked LOH as a result of CIN when genome-wide or specific regions such as chromosomes 12p, 14q, and 17q were investigated. Moreover, LOH at 3p loci containing genes associated with antioxidant defenses (e.g., glutathione peroxidase I) is not only associated with the development of lung cancer but also with higher responsiveness to DNA damaging agents (e.g., radiation).
Multiple mechanisms during cell cycle progression have been implicated in the advent of CIN and aneuploidy in lung cancer. These include failure at the mitotic checkpoint, mutations and amplifications in the kinetochore (protein structure on chromatids where the spindle fibers attach during cell division) and centrosome components, and mutations in DNA repair genes. The mitotic checkpoint, also called the spindle assembly checkpoint, is activated when the kinetochore is not attached to the spindle, lacks microtubules, or has poor or inadequate tension, thereby deregulating metaphase–anaphase progression. Loss-of-function mutations, or reduced gene expression of the mitotic checkpoint genes (mitotic arrest deficient-like 1 [ MAD1 / MAD2 ] and mitotic checkpoint serine/threonine kinase [ BUB1 , BUBR1 ]), lead to chromosomal missegregation and contribute to aneuploidy. Given that loss of mutations in mitotic checkpoint genes were rarely detected in lung cancers (less than 3%) in the recent comprehensive genome-wide sequencing data collection, development of lung cancer and CIN is more closely related to their dysfunction due to phosphorylation or cytoplasmic location. Interestingly, a study in MAD2 +/– p53 +/– and MAD1 +/– MAD2 +/– p53 +/– mice suggested a cooperative role of MAD1 / MAD2 and p53 genes in generating increased aneuploidy and tumorigenesis. Furthermore, the mitotic checkpoint has also been linked to DNA-damage response, and a defective mitotic checkpoint confers cancer cells’ resistance to certain DNA-damaging anticancer drugs. The centrosomes are thought to maintain genomic stability through the establishment of bipolar spindles during cell division, ensuring equal segregation of replicated chromosomes to two daughter cells. STK15, encoding aurora kinase A (AURKA), is amplified and overexpressed in diverse types of human tumors, leading to centrosome amplification, CIN, and tumorigenesis. Aurora kinases are serine/threonine kinases that function as key regulators of the mitosis process. Their dysfunction interferes with cell cycle checkpoints and allows genetically aberrant cells to enter mitosis and undergo cell division. Overexpression of aurora kinases can lead to aneuploidy, resulting in the failure to maintain chromosomal integrity.
In one study, AURKA was highly overexpressed in 50% of NSCLC, and its overexpression was significantly upregulated in tumor samples compared with matched lung tissue ( p < 0.01), suggesting a role as a tumor marker. Moreover, AURKA was principally upregulated in moderately and poorly differentiated lung cancers, as well as in SCCs and adenocarcinomas, compared with the noninvasive bronchioloalveolar subtype. The frequency of AURKA amplification in NSCLC ranges from 1% to 6% and seems to be more common in lung adenocarcinomas than in lung SCCs. In comparison, aurora kinase B (AURKB) plays a less clear role in tumorigenesis. However, many studies now support an association between AURKB and malignant transformation, with the involvement of additional factors. Although AURKB overexpression alone did not transform rodent fibroblast cells, increased kinase activity did facilitate Harvey rat sarcoma viral oncogene homolog ( HRAS )-induced transformation, which led to the production of aneuploid cells. In an IHC analysis of 160 NSCLC samples, 78% of tumors were found to overexpress AURKB, and its overexpression was also associated with adverse tumor features and poor prognosis in lung adenocarcinomas. Contrary to AURKA, the overexpression of which is associated with gene amplification, AURKB overexpression was associated with aberrant transcriptional regulation in primary lung carcinoma.
Therefore overexpression and amplification of aurora kinases have been associated with neoplastic transformation, serving as attractive targets for cancer therapy. A growing number of inhibitors of aurora kinases have been developed and, at the time of publication, were being evaluated in clinical trials to assess the therapeutic potential of aurora-based targeted therapy. These inhibitors include AMG900 (Amgen, Thousand Oaks, CA, USA), AT9283 (Astex Therapeutics, Dublin, CA, USA), AZD1152 (Astra Zeneca, London, UK), and PF03814735 and BI811283 (Boehringer-Ingelheim, Ridgefield, CT, USA). Some of these drugs have selective activity against one aurora kinase subtype, whereas others exhibit pan-inhibitory effects.
In addition to mitotic checkpoint proteins and centrosome components, CIN may be caused by defects in the DNA double-strand break repair genes—ataxia telangiectasia mutated ( ATM ), BRCA1 , BRCA2 , x-ray repair complementing defective repair in Chinese hamster cells (double-strand-break rejoining; XRCC5 )—or DNA-damage response. Interestingly, the chromosomal regions at 2q33–35 and 13q12.3, which included loci encoding the XRCC5 and BRCA2 genes, showed a high frequency of LOH in NSCLC. More recently, it was reported that low messenger RNA and protein expressions in BRCA1 / BRCA2 and XRCC5 genes occur in lung adenocarcinoma and SCC and that promoter hypermethylation is the predominant mechanism in deregulation of these genes. Given that BRCA1/BRCA2 proteins are central to p53-dependent elimination of tetraploid or aneuploid (often preceded by tetraploid state) cells, it is not surprising that these proteins are frequently inactivated or downregulated in NSCLC, synergizing with p53 inactivation to establish an atmosphere of tolerance for a nondiploid state. Although unrepaired or incorrectly repaired DNA lesions may give rise to cancer-initiating mutations, one way to efficiently tackle cancer is to take advantage of such biologic differences between cancer and normal cells and exploit the defects of tumor-associated DNA-damage response in smart therapeutic strategies.
Amplification as a Mechanism of Oncogenesis
Gene amplification refers to the expansion of gene copy number in a restricted region of a chromosome arm. It is prevalent in some tumors and is often associated with overexpression of the amplified gene, causing cancer cells to grow or become resistant to anticancer drugs. Often, although not necessarily, gene amplification is seen as karyotypic abnormalities including the extrachromosomal, acentric structure known as double minutes and the homogeneously staining regions ( Fig. 10.1 ). High-throughput genomic analyses of thousands of cancer specimens showed that the majority (approximately 75%) of the gene amplifications were focal in nature (50 kb to 300 kb) and targeted primarily oncogenes, encoding signaling proteins crucial for cellular proliferation and survival. This finding strongly supports the notion that gene amplification promotes tumor formation, tumor maintenance, and drug resistance. The preponderance of focal amplification targeting oncogenes contrasts sharply with large genomic deletions, which are mostly passenger mutations with only a few exceptions, such as cyclin-dependent kinase inhibitor 2A/B ( CDKN2A/B ), retinoblastoma 1 ( RB1 ), and FAT atypical cadherin 1 ( FAT1 ) tumor suppressor genes. The contributing factors to CIN, including common chromosomal fragile sites, errors in DNA replication, and telomere dysfunction, are causally linked to amplification and large genomic deletions.
Gene amplification is a common mechanism of oncogenic activation in NSCLC and, on a whole genome scale, has a strong effect on the level of protein expression. Given the potential role of gene amplification in lung tumorigenesis and tumor progression, this event is commonly associated with unique clinicopathologic features and aggressive tumor behavior. Not surprisingly, amplifications of the EGFR, v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 ( ERBB2 ), met proto-oncogene ( MET ), MYC , and fibroblast growth factor receptor 1 ( FGFR1 ) genes have been reported to be significantly associated with poor prognosis in NSCLC (as will be discussed). In addition, amplification has been identified as a mechanism of resistance to therapy.
Because the tumor can become dependent on overexpression of the oncogenes for its survival and proliferation, amplifications of oncogenes usually define unique subsets of lung cancer and support their use as therapeutic targets. As best illustrated by the example of the success of trastuzumab in ERBB2-amplified breast cancer, amplification of specific oncogenes may provide diagnostic utility based on their impact on therapeutic response and patient outcome. In lung cancer, however, although the results of several studies have clearly demonstrated the clinical usefulness of testing for EGFR mutations and anaplastic lymphoma kinase (ALK) fusions to guide treatment and improve patient outcomes, selection of therapy based on gene amplification is yet to be approved.
This chapter focuses on the most relevant examples of gene amplifications that have shown diagnostic usefulness because of prognostic and predictive values.
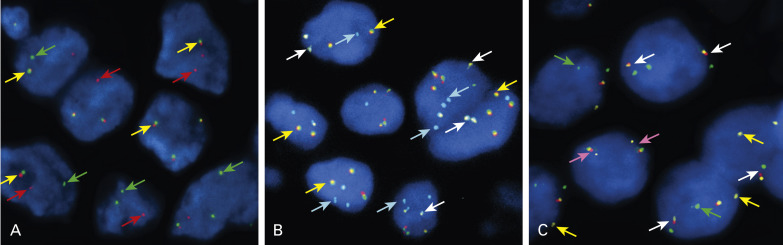
EGFR Amplification
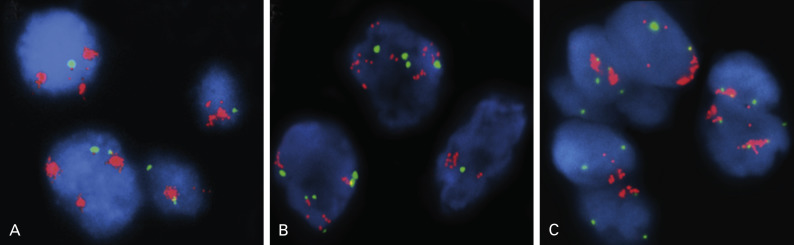
Determination of EGFR gene copy number by fluorescence in situ hybridization (FISH) has prognostic and diagnostic usefulness in NSCLC ( Fig. 10.1A ). Based on a retrospective study of patients with advanced NSCLC who were treated with gefitinib, tumors are considered to have a high EGFR gene copy number (EGFR FISH+) if they show a high copy number or amplification of the EGFR gene. Overall, tumors with a high EGFR gene copy number represent approximately 30% of NSCLC. Unlike EGFR mutations, which are more frequently found in Asian, female, and never-smoking patients, and in patients with adenocarcinoma histology, the distribution of EGFR copy numbers is mostly independent of these clinicopathologic characteristics. Lung cancers with a high EGFR gene copy number seem to be associated with a worse prognosis than cancers with a low EGFR gene copy number, in both early and advanced stages. EGFR amplification usually coexists with mutations in lung adenocarcinoma, suggesting that the mutation occurs first, then induces gene amplification during tumor progression and metastasis. This hypothesis was clearly illustrated in a biomarker analysis of Iressa Pan-Asia Study (IPASS) that showed a concordance between EGFR mutation and high gene copy number in almost 90% of patients. Interestingly, in an analysis from the Iressa Survival Evaluation in Lung Cancer phase III study in which patients were predominantly of non-Asian origin, the concordance rate between these two biomarkers seemed much lower, suggesting that the mechanism of the genomic gain of EGFR may have ethnic differences.
Although EGFR gene copy number has been evaluated as a predictive biomarker for sensitivity to EGFR tyrosine kinase inhibitors (TKIs) in several studies, its predictive role remains controversial. Early studies showed that patients with EGFR FISH+ tumors were most likely to benefit from treatment with EGFR TKIs. However, in IPASS, patients with tumors with a high EGFR gene copy number had significantly longer progression-free survival with the EGFR inhibitor gefitinib only in the presence of an EGFR mutation, whereas patients with an EGFR mutation had longer progression-free survival with gefitinib irrespective of the EGFR gene copy number. These findings suggest that the predictive value of the EGFR amplification was driven by coexisting EGFR mutations. The robust predictive value of EGFR mutations has been confirmed in subsequent phase III studies comparing first-line EGFR TKIs with chemotherapy in advanced NSCLC with activating EGFR mutations.
Nevertheless, similar to the case of EGFR mutations, amplification of the EGFR gene can fully activate EGFR tyrosine kinase and trigger downstream oncogenic pathways. Therefore it seems reasonable to assume a correlation between an abnormality in the EGFR copy number and EGFR TKI sensitivity. In support of this hypothesis, it was reported that a high EGFR gene copy number may be used as a predictive marker in patients with advanced squamous cell lung carcinoma, in which activating EGFR mutations are very rare. In addition to its effect on the prognosis and response to EGFR TKIs, focal amplification of EGFR that preferentially involves the T790M-containing allele confers resistance to the irreversible EGFR TKIs (e.g., dacomitinib).
ERBB2 Amplification
ERBB2 is also a member of the family of EGFR tyrosine kinases, but it is not activated by a known cognate ligand and instead serves as a preferred dimerization partner to other family members. Amplification of the ERBB2 gene, mapped at 17q11.2–q12, was reported to occur in approximately 2% of unselected NSCLCs, with a rise in frequency to 11% in poorly differentiated adenocarcinomas. It was also reported that approximately 40% of ERBB2-amplified tumors had concurrent amplification and/or mutation of EGFR. Therefore it is not surprising that ERBB2 amplification ( Fig. 10.1B ) was more frequently associated with female gender and never-smoking status, characteristics that are associated with the presence of EGFR mutation and amplification. ERBB2 amplification correlates well with protein overexpression and is associated with higher tumor grade, higher disease stage, and shorter survival, all of which provide evidence supporting this receptor as a useful molecular target in the treatment of NSCLC. Unfortunately, the addition of trastuzumab to gemcitabine and cisplatin did not appear to provide any benefit for patients with advanced NSCLC with ERBB2 overexpression or amplification. However, because very few patients had ERBB2 3+/ amplification in this trial, further evaluation of trastuzumab in this specific subset of patients with lung cancer is desirable. Given the high intratumoral heterogeneity of ERBB2 amplification and discrepancies between primary tumors and their metastases, careful testing should be considered in the assessment of patients with NSCLC as candidates for ERBB2-targeted therapy.
Of note, a high ERBB2 gene copy number (ERBB2/FISH+) had a positive additive effect on the efficacy of EGFR TKI in the presence of EGFR mutation and/or amplifications, suggesting that testing of the ERBB2 gene copy number may have a complementary role for selection of patients who gain the greatest benefit from EGFR TKIs. By contrast, ERBB2 amplification is an example of an acquired resistance mechanism to the EGFR inhibitor cetuximab. In the HCC827 NSCLC cell, aberrant ERBB2 activation leads to persistent extracellular signal-regulated kinase 1/2 signaling in the presence of cetuximab, thus preventing cetuximab-mediated growth inhibition.
MET Amplification
MET, a proto-oncogene located on 7q31, encodes a transmembrane tyrosine kinase receptor for hepatocyte growth factor (HGF). Binding of HGF to MET induces receptor dimerization and transphosphorylation, triggering conformational changes that activate MET tyrosine kinase activity. Preclinical findings also suggest that lung cancer cell lines harboring MET gene amplification are dependent on MET for growth and survival. HGF stimulation of MET gene amplification leads to the activation of a number of signaling pathways, including phosphatidylinositol 3-kinase/v-akt murine thymoma (PI3K)/AKT, RAS/mitogen-activated protein kinase (MAPK), and phospholipase C-γ pathways.
The frequency of MET amplification is rare and has been reported to be 1.4% to 7.3% among patients with NSCLC not previously treated with EGFR TKIs. MET gene amplification, or MET FISH+ ( Fig. 10.1C ), has not been associated with gender, histology, or smoking status, but has been significantly associated with higher tumor grade and advanced stage. Interestingly, albeit mutually exclusive with mutations in EGFR , ERBB2 , and KRAS genes, MET FISH+ status was significantly associated with EGFR FISH+ status, likely because both genes are located in chromosome 7. This finding may support the early preclinical demonstration of interaction between EGFR and MET signaling pathways. For patients who had surgical resection, survival was shorter for those with MET FISH+ tumors (5 or more copies per cell) than for those with MET FISH– tumors.
The rarity of MET amplification in NSCLC, particularly at the high levels found in EGFR TKI–resistant cell line models (MET gene copy number greater than 12), suggested that this event plays a limited role in primary resistance to EGFR TKIs. Instead, HGF-stimulated MET signaling activation is more likely to be responsible for primary resistance to EGFR TKIs. Autocrine or paracrine secretion of HGF results in MET activation, reactivation of the MAPK, and PI3K/AKT signaling pathways and immediate resistance to EGFR inhibition. Indeed, HGF and MET have been reported to participate in paracrine tumorigenic pathways in several other malignancies.
By contrast, MET amplification is present in approximately 20% of tumors with acquired resistance to EGFR TKIs. Engelman et al. reported that NSCLC overcomes inhibition of EGFR TKIs by amplifying the MET oncogene to activate ERBB3, a member of the EGFR family, and the PI3K/AKT cell survival pathway. In another study, Bean et al. showed MET amplification in 21% of patients with acquired resistance to gefitinib or erlotinib and in only 3% of untreated patients, confirming that MET could be a relevant therapeutic target for some individuals with acquired resistance to EGFR TKIs.
PIK3CA Amplification
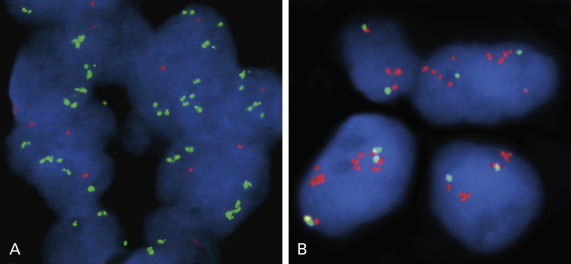
PI3K signaling is a major oncogenic pathway that functions in cancer cell growth, survival, motility, and metabolism. Amplification of the phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha ( PIK3CA gene), mapped at 3q26.3 and encoding the p110 catalytic subunit, was more frequently found in men, smokers, and patients with SCC. Overall, the incidence of PIK3CA gene amplification has been reported to be 33.1% to 70% in squamous cell lung carcinoma and 1.6% to 19% in lung adenocarcinoma, suggesting that this genetic alteration mainly targets squamous cell lung carcinoma. Furthermore, a high level of PIK3CA copy gain was present exclusively in SCCs. Amplification of PIK3CA ( Fig. 10.2A ) occurs at higher frequencies than genomic mutations in lung cancer and they occur independently of each other, implying that either molecular event has equivalent oncogenic potential.
The functional importance of PIK3CA gene amplification is shown by increased PI3K activity and phosphorylated AKT. Knockdown of PIK3CA inhibits anchorage-dependent and anchorage-independent growth in PIK3CA-amplified NSCLC cells, but has no effect in cells harboring wild-type PIK3CA. Interestingly, coexistence of PIK3CA gene amplification and EGFR or KRAS mutation in a single tumor was less frequent in SCC than in adenocarcinoma. This finding suggests that PIK3CA copy gain may play a pivotal role in pathogenesis of lung SCCs, further providing a rationale for targeting the PI3K pathway in this disease. The significance of the PI3K pathway as a therapeutic target in squamous cell lung carcinomas has been highlighted in the most recent study by the Cancer Genome Atlas Research Network. In that study, alterations in the PI3K/AKT pathway were found in 47% of tumors and, more important, approximately 38.2% of tumors (68 of 178) had PIK3CA amplifications. This finding is of particular interest because targeted agents have been successful for the treatment of only lung adenocarcinoma. The functional dependence of squamous cell lung carcinoma on the PI3K pathway should be validated by the successful treatment with targeted PI3K inhibitors, and such trials are underway.
FGFR1 Amplification
The FGFR tyrosine kinase family comprises four kinases (FGFR 1–4) and plays a crucial role in cancer cell growth, survival, and resistance to chemotherapy. Amplification of the FGFR1 locus at chromosome 8p12 has been described in several tumor types, particularly SCC of the lung ( Fig. 10.2B ) and SCLC.
FGFR1 gene amplification is more commonly found in squamous cell lung carcinoma than lung adenocarcinoma, with a relatively high incidence of up to 24.8%, and has recently been reported to be a novel druggable target in this specific histologic subset. Interestingly, Kim et al. reported that the incidence of FGFR1 amplification was also associated with smoking status in a dose-dependent manner (current smoker, 28.9% vs. former smoker, 2.5% vs. nonsmoker, 0%; p < 0.0001), suggesting that FGFR1 gene amplification is an oncogenic aberration caused by cigarette smoking. FGFR1 gene amplification drives downstream activation of PI3K/AKT and RAS/MAPK signaling, and a selective FGFR inhibitor caused downstream inhibition and induction of apoptosis in FGFR1-amplified squamous cell lung carcinomas, strongly supporting the utility of FGFR1 gene amplification as a relevant therapeutic target in this disease.
FGFR1 gene amplification also has been reported to hold a significant prognostic value. Kim et al. found that FGFR1 amplification is a negative prognostic factor for patients with resected SCC of the lung, whereas Heist et al. reported there was no significant difference in overall survival by FGFR1 amplification status. The conflicting results may be related to differences in method and cutoff values used to assess and define FGFR1 amplification.
In one study, FGFR1 amplification was noted in 5.6% of SCLC, mostly at high levels and as homogeneous staining regions. Furthermore, inhibition of FGFR has resulted in a blockade of tumor growth, suggesting an important role of the FGF–FGFR signaling pathway for SCLC growth, which indicates that FGFR1 amplification may also be a therapeutic target in SCLC.
Standardized screening criteria have been proposed to reliably identify patients who have lung cancers with FGFR1 amplifications for clinical trials with FGFR inhibitors. According to these criteria, high-level FGFR1 amplification is defined as an FGFR1/centromere 8 ratio of 2.0 or higher, an average number of FGFR1 signals per tumor cell nucleus of six or more, or 10% or more tumor cells containing at least 15 FGFR1 signals or large clusters; low-level amplification is defined as five or more FGFR1 signals in at least 50% of tumor cells. The utility of the proposed criteria should be validated by the clinical response data from clinical trials with FGFR inhibitors.
The identification of FGFR1 amplification holds promise for the development of novel molecularly targeted therapeutic agents in the treatment of squamous cell lung carcinoma and SCLC, and recently developed FGF/FGFR-targeting anticancer agents are being studied in clinical trials.
Structural Changes Leading to Oncogenesis by Gene Fusions
ALK Fusion
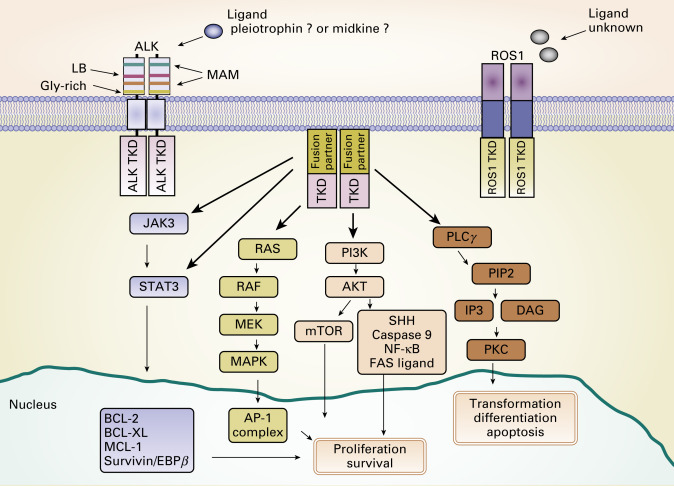
In 2007, Soda et al. discovered that oncogenic fusion genes consisting of echinoderm microtubule-associated protein like 4 ( EML4 ) and ALK are present in a small subset of NSCLCs. The endogenous ALK gene is normally not expressed in most adult tissues, including the lung epithelium, but EML4–ALK fusion leads to both ectopic expression and constitutive activation of ALK and its downstream signaling pathways, resulting in uncontrolled cellular proliferation and survival ( Fig. 10.3 ). The prevalence of ALK fusions has been reported to be approximately 4% (range, 1.5% to 7.5%) in unselected NSCLC populations, representing potentially 40,000 new cases worldwide each year. The frequency of ALK-positive tumors in NSCLC and methods to detect ALK have been evaluated in population studies ( Table 10.1 ). ALK-positive lung cancers are highly sensitive to ALK inhibitors, and an understanding of the resistance mechanisms to ALK inhibitors is crucial for the optimal therapy of ALK-positive NSCLC.
| Study | Study Population (No. of patients) | No. of ALK Fusions (%) | Detection Method | Fusion Variants |
|---|---|---|---|---|
| Soda et al. | Japanese (75) | 5 (6.7) | RT-PCR | EML4–ALK (E13; A20, E20; A20) |
| Takeuchi et al. | Japanese (364) | 11 (3.0) | 10RT-PCR | EML4–ALK (E13; A20, E20; A20) |
| Wong et al. | Chinese (266) | 13 (5.0) | RT-PCR Direct sequencing | EML4–ALK (E6; A20, E13; A20, E20; A20, E18; A20) |
| Inamura et al. | Japanese (221) | 5 (2.0) | RT-PCR | EML4–ALK (E20; A20) |
| Shinmura et al. | Japanese (77) | 2 (3.0) | RT-PCR | EML4–ALK (E13; A20, E20; A20) |
| Koivunen et al. | Korean/U.S. (305) | 8 (3.0) | RT-PCR | EML4–ALK (E13; A20, E20; A20, E6a/b; A20, E15; A20) |
| Shaw et al. | Predominantly white (141) a | 19 (13.0) | FISH | NA |
| Kim et al. | Korean (229) a | 19 (8.3) | FISH | NA |
| Gainor et al. | White/Asian (1683) | 75 (4.4) | FISH | NA |
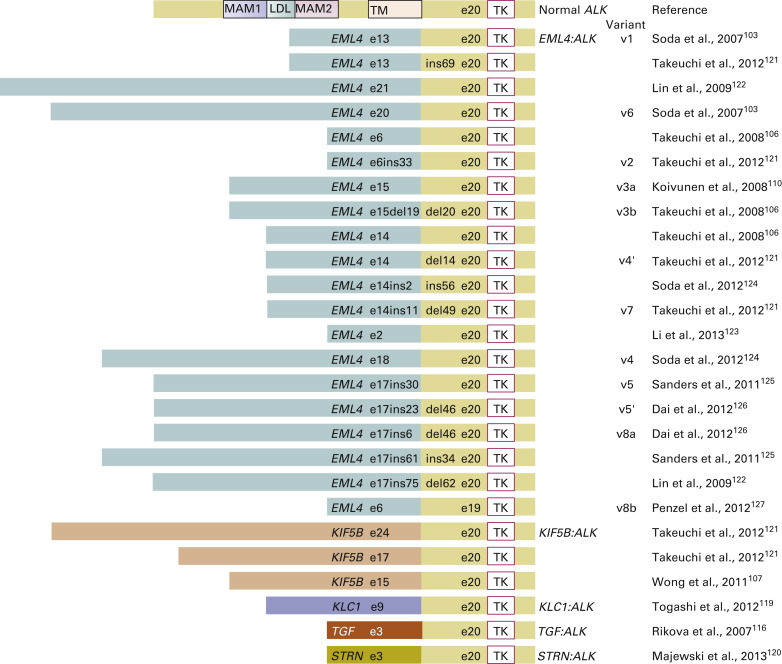
ALK fusions result from various types of chromosomal rearrangements, all of which lead to aberrant activation of ALK. EML4–ALK fusion, the most frequent ALK fusion in NSCLC, is the result of paracentric inversions (intrachromosomal rearrangement) involving the short arm of chromosome 2. Multiple studies have shown other, rare, fusion partners of ALK , such as kinesin family member 5B ( KIF5B ; 10p11.22), TRK-fused gene ( TFG ; 3q12.2), kinesin light chain 1 ( KLC1 ; 14q32.3), and striatin, calmodulin binding protein ( STRN ; 2p22.2). ALK fusion with KIF5B , TFG , or KLC1 is the result of interchromosomal rearrangement, whereas ALK fusion with STRN is the result of intrachromosomal deletion. Despite the diversity of fusion partners, most of them contain coiled-coil or leucine zipper domains that drive the dimerization or oligomerization of fusion kinase, which leads to ligand-independent activation of the tyrosine kinase. To date, more than 20 EML4–ALK variants have been identified in NSCLC ( Fig. 10.4 ). Despite variable breakpoints of EML4 (exons 2, 6, 13, 14, 15, 17, 18, 20, and 21), the genomic breakpoint within the ALK gene is conserved at exon 20 with few exceptions at exon 19. Therefore all EML4–ALK fusion proteins involve the intracellular tyrosine kinase domain of ALK. All those rearrangements are potentially detected by the break-apart FISH probe (Vysis ALK Break-Apart FISH Probe Kit, Abbott Molecular, Abbott Park, IL, USA; Fig. 10.5A ). It is still unknown whether any particular EML4–ALK fusion variant may confer differential sensitivity to ALK inhibitors, which may underlie the heterogeneity in responses among patients with ALK-positive NSCLC. Heuckmann et al. reported that EML4–ALK v2 had the shortest half-life and greatest sensitivity to crizotinib, whereas v1 and v3b had intermediate sensitivity and v3a had the least sensitivity.
ALK fusion has been associated with several distinct clinicopathologic features and treatment outcomes. A strong association between ALK fusions and a never-smoking or light-smoking (fewer than 10 pack-years) history has been reported, with the frequency of ALK fusions in the never-smoking or light-smoking subgroup higher than in the unselected population and ranging from 8.3% to 39%. A younger age at diagnosis and adenocarcinoma histology are other important features associated with ALK -positive lung cancers, and ALK fusions rarely overlap with other oncogenic drivers. Furthermore, ALK -positive tumors are substantially more likely to have abundant signet ring cells. Contrary to EGFR -mutant NSCLC, ALK -positive NSCLC has shown resistance to EGFR TKIs, and sensitivity of ALK -positive NSCLC to platinum-based chemotherapy has not differed from that of ALK -negative NSCLC. The activity of pemetrexed-based chemotherapy in ALK -positive NSCLC is controversial and needs further validation. According to guidelines from the National Comprehensive Cancer Network and from the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology, testing of ALK fusion and EGFR mutation is now recommended for all patients with advanced nonsquamous NSCLC. Therefore given the low frequency of this genetic alteration, efficient screening for ALK fusion is a crucial issue in clinical practice. Currently, reverse transcription (RT)-PCR, FISH, and IHC have been used to detect ALK fusion. RT-PCR is a potentially rapid diagnostic method with high sensitivity. It provides direct evidence of the genomic fusion, but the difficulty in obtaining high-quality RNA limits the clinical utility of this method. FISH is currently the standard criterion used in clinical trials for detection of ALK fusion, and it was the first Food and Drug Administration (FDA)-approved method (Vysis ALK Break-Apart FISH Probe Kit) for use of crizotinib in ALK -positive NSCLC. Any type of ALK fusions could theoretically be detected using this method, but the main disadvantages are a relatively high cost and the specialized technical training required. Because normal adult tissue, except for neural tissue, does not express ALK , IHC has been reported to be quite effective at detecting ALK fusion in several studies. The sensitivity of ALK IHC is highly dependent on the affinity of the primary antibody and the signal amplification system. Using high-affinity antibody clones and a sensitive detection system, the overall sensitivity and specificity of ALK IHC were 90% to 100% and 95.2% to 98.0%, respectively. However, methodologic standardization and proof of clinical utility of the ALK IHC assay are still in progress.
Crizotinib has shown significant clinical benefit in ALK -positive NSCLC. During phase I (PROFILE 1001) and phase II (PROFILE 1005) studies of crizotinib, the objective response rate was approximately 60%. The responses were often rapid and durable, and the median duration of response was 49.1 weeks; the median progression-free survival was 9.7 months in the most recent update of the phase I study. Crizotinib has been well tolerated, with mild adverse events, including visual disturbance, nausea/vomiting, diarrhea, constipation, and peripheral edema. On the basis of clinical activity and tolerability demonstrated in phase I and phase II studies, crizotinib received accelerated FDA approval in August 2011 for the treatment of advanced ALK -positive NSCLC. This approval was conditioned on the results of randomized studies comparing crizotinib with standard chemotherapy (PROFILE 1007 and 1014). The results from the PROFILE 1007 phase III study were reported in 2013. In this study, patients with advanced ALK -positive NSCLC were randomly assigned to receive either crizotinib or standard second-line chemotherapy (pemetrexed or docetaxel). Crizotinib was significantly superior to standard chemotherapy in terms of overall response (65% vs. 19%; p < 0.001) and progression-free survival (7.7 months vs. 3 months; p < 0.001). These results led to the regular approval by the US FDA in November 2013 for the treatment of advanced ALK -positive NSCLC.
Given the early success of crizotinib for the treatment of ALK -positive NSCLC, many next-generation ALK inhibitors are in development ( Table 10.2 ). Some of these newer ALK inhibitors have shown activity against mutant forms of ALK that are resistant to crizotinib.
| Drug | Sponsor a | Phase of Trial | Primary End Point | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| AP26113 | Ariad | I/II | Overall response rate | 01449461 |
| CH5424802 | Hoffmann-La Roche | I I/II (crizotinib-naive) I/II (crizotinib-failed) | Recommended phase II dose Overall response rate | 01588028 01871805 01801111 |
| PF-06463922 | Pfizer | I/II | Dose-limiting toxicity Overall response rate | 01970865 |
| Ganetespib | Synta | II | Overall response rate | 01562015 |
| AUY922 | Massachusetts General Hospital | II | Overall response rate Overall response rate Progression-free survival Progression-free survival | 01752400 |
| LDK378 | Novartis | II (crizotinib-naive) II (crizotinib-failed) III III | RT-PCR | 01685138 01685060 01828099 01828112 |
| X-396 | Xcovery | I | Maximum tolerated dose | 01625234 |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


