INTRODUCTION
SUMMARY
The blood coagulation system, like a powerful idling engine, is always active and generating thrombin at very low levels, poised for explosive thrombin generation. Positive feedback activation of factors V, VII, VIII, and XI imparts special threshold properties to blood coagulation, making the coagulant response nonlinearly responsive to stimuli. Overt blood coagulation represents a threshold system with apparent all-or-none responses to various levels of stimuli, and an ensemble of opposing reactions determines the ultimate upregulation and downregulation of thrombin generation both locally and systemically. Cellular and humoral anticoagulant mechanisms synergize with plasma coagulation inhibitors to prevent massive thrombin generation in the absence of a substantial procoagulant stimulus. This chapter highlights mechanisms that inhibit blood coagulation, with an emphasis on defects of plasma proteins that cause hereditary thrombophilias. Major thrombophilic defects involve the anticoagulant protein C pathway, comprising multiple cofactors or effectors that additionally include thrombomodulin, endothelial protein C receptor, protein S, high-density lipoprotein, and factor V. Activated protein C exerts multiple protective homeostatic actions, including proteolytic inactivation of factors Va and VIIIa, as well as direct cell-signaling activities involving protease activated receptors 1 and 3, endothelial cell protein C receptor, integrin CD11b/CD18, and apolipoprotein E receptor 2. The factor V Leiden variant causes hereditary activated protein C resistance by impairing the ability of the protein C pathway to inhibit coagulation because it cannot properly cleave factor Va Leiden. Plasma protease inhibitors are also key to block coagulation. Antithrombin inhibits thrombin and factors Xa, IXa, XIa, and XIIa, in reactions stimulated by physiologic heparan sulfate or pharmacologic heparins. Tissue factor pathway inhibitor neutralizes the extrinsic coagulation pathway factors VIIa and Xa. Other plasma protease inhibitors can also neutralize various coagulation proteases.
Control of coagulation reactions is essential for normal hemostasis. As part of the tangled web of host defense systems that respond to vascular injury, the blood coagulation factors (Chap. 113) act in concert with the endothelium and blood cells, especially platelets, to generate a protective fibrin-platelet clot, forming a hemostatic plug. Pathologic thrombosis occurs when the protective clot is extended beyond its beneficial size, when a clot occurs inappropriately at sites of vascular disease, or when a clot embolizes to other sites in the circulatory bed. For normal hemostasis, both procoagulant and anticoagulant factors must interact with the vascular components and cell surfaces, including the vessel wall (Chap. 115) and platelets (Chap. 112). Moreover, the action of the fibrinolytic system must be integrated with coagulation reactions for timely formation and dissolution of blood clots (Chap. 135). This chapter on control of coagulation highlights the major physiologic mechanisms for downregulation of blood coagulation reactions and the plasma proteins that inhibit blood coagulation, with an emphasis on those mechanisms whose defects are clinically significant based on insights gleaned from consideration of the hereditary thrombophilias (Chap. 130). Chapter 113 provides a complete description of blood coagulation factors and hemostatic pathways.
Acronyms and Abbreviations
APC, activated protein C; apoER2, apolipoprotein E receptor 2; CD11b/CD18, Mac-1; EPCR, endothelial cell protein C receptor; GLA, γ-carboxyglutamic acid; GPI, glycosylphosphatidylinositol; HDL, high-density lipoprotein; NMDA, N-methyl-d-aspartate; PAR-1, protease-activated receptor-1; serpin, serine protease inhibitor; SHBG, sex hormone–binding globulin; TFPI, tissue factor pathway inhibitor; TNF, tumor necrosis factor; ZPI, protein Z–dependent protease inhibitor.
BLOOD COAGULATION PATHWAYS AND THE PROTEIN C PATHWAYS
Although decades have elapsed since the elaboration of the cascade model1,2 for blood coagulation (see Chap. 113, Fig. 113–27), the basic outline of sequential conversions of protease zymogens to active serineproteases is still useful, albeit with important modifications (see Chap. 113, Fig. 113–28), to represent blood coagulation reactions. The major conceptual advances for procoagulant pathways in the past two decades emphasize both positive and negative feedback reactions affecting thrombin generation as depicted in Fig. 114–1.
Figure 114–1.
Blood coagulation pathways and protein C anticoagulant pathway. Thrombin can be either a procoagulant (left) or an anticoagulant (right), depending on cofactors and surfaces. Coagulant thrombin clots fibrinogen and activates platelets and factors V, VIII, XI, and XIII. Conversion of zymogen protein C to the active protease, APC, by thrombomodulin-bound thrombin is enhanced by endothelial protein C receptor (EPCR). APC with its nonenzymatic cofactor, protein S, inactivates factors Va and VIIIa by highly selective proteolysis (e.g., at Arg506 and Arg306 in factor Va), yielding inactivated (i) factors Vi and VIIIi. This anticoagulant action may be enhanced by phospholipid (PhosLipid) surfaces on platelets, endothelial cells, or their microparticles. High-density lipoprotein (HDL) can also provide protein S–dependent anticoagulant APC-cofactor activity. Similarly, neutral glycosphingolipids such as glucosylceramide (GLcCer) can enhance APC anticoagulant activity. GPIb, glycoprotein Ib; PAR, protease-activated receptor. (Adapted with permission from Griffin JH: Blood coagulation. The thrombin paradox. Nature 378(6555):337–338, 1995.)
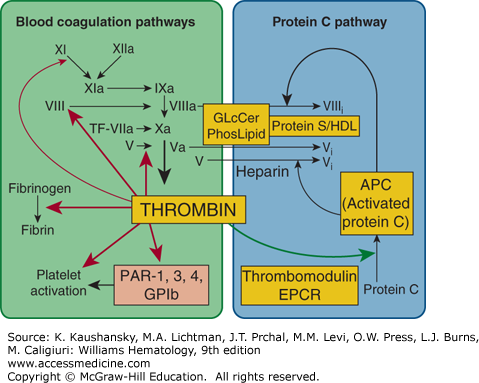
In positive feedback reactions, procoagulant thrombin activates platelets and factors V, VIII, and XI (Chap. 113).3,4,5 Small amounts of thrombin can be generated by trace amounts of tissue factor via the extrinsic pathway. Subsequently, thrombin can activate factors XI, VIII, and V, thereby stimulating each of the steps in the intrinsic pathway, thereby amplifying thrombin generation (see Fig. 114–1).
In negative feedback reactions, anticoagulant activated protein C (APC) that is generated on endothelial cell surfaces6,7,8 (Fig. 114–2) downregulates coagulation (see Figs. 114–1 and 114–3). Furthermore, APC can exert direct cytoprotective effects on cells via reactions that involve certain receptors, including endothelial cell protein C receptor (EPCR) and protease-activated receptor-1 (PAR-1) (Fig. 114–4), PAR-3, integrin CD11b/CD18, and possibly apolipoprotein E receptor 2 (apoER2).7,8 APC’s cytoprotective effects include antiinflammatory and antiapoptotic activities, as well as alterations of gene-expression profiles and stabilization of endothelial barriers (see “Activated Protein C Activities” below). Because inflammation, apoptosis, and vascular barrier breakdown contribute significantly to reactions that promote thrombin generation, such direct cytoprotective effects of APC on cells indirectly downregulate thrombin generation.7,8
Figure 114–2.
Protein C activation on endothelial cell surface. On an endothelial surface, activated protein C (APC) generation follows binding of protein C (PC) to endothelial protein C receptor (EPCR) where PC is activated by limited proteolysis by the thrombin (IIa)–thrombomodulin (TM) complex. This action of thrombin liberates a dodecapeptide (residues 158 to 169) from protein C to generate the multifunctional protease APC. (Adapted with permission from Mosnier LO, Zlokovic BV, Griffin JH: The cytoprotective protein C pathway. Blood 109(8):3161–3172, 2007.)
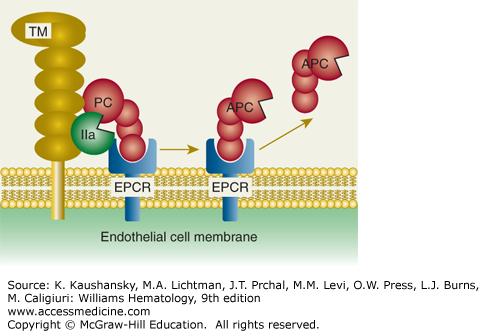
Figure 114–3.
Activated protein C (APC) exerts its anticoagulant activity by proteolytic inactivation of factors Va and VIIIa on membrane surfaces containing phospholipids that are derived from cells, lipoproteins, or cellular microparticles. A variety of lipid and protein cofactors (see Fig. 114–1 legend and text) accelerate the inactivation of factors Va and VIIIa to yield the irreversibly inactivated factors Vi and VIIIi. (Adapted with permission from Mosnier LO, Zlokovic BV, Griffin JH: The cytoprotective protein C pathway. Blood 109(8):3161–3172, 2007.)
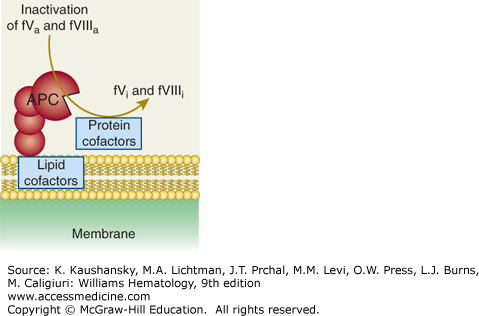
Figure 114–4.
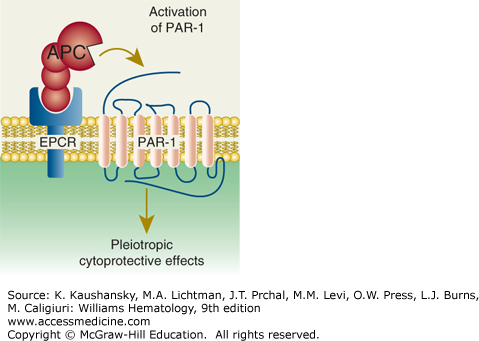
Paradigm for activated protein C (APC)’s initiation of cell signaling and multiple cytoprotective effects. Direct effects of APC on cells are initiated by activation of the G-protein–coupled receptor, protease-activated receptor-1 (PAR-1), by endothelial protein C receptor (EPCR)-bound APC. The γ-carboxyglutamic acid (GLA) domain of APC binds to EPCR to help position APC’s protease domain for efficient cleavage of the extracellular N-terminal tail of PAR-1, which results in G-protein–coupled receptor activation and subsequent antiinflammatory and antiapoptotic effects, alterations of gene expression profiles, and stabilization of endothelial junctions. (Adapted with permission from Mosnier LO, Zlokovic BV, Griffin JH: The cytoprotective protein C pathway. Blood 109(8):3161–3172, 2007.)
For APC generation by the protein C cellular pathway, binding of thrombin to thrombomodulin converts the bound thrombin from a procoagulant enzyme to an anticoagulant enzyme that converts the protein C zymogen to an anticoagulant serine protease, APC (see Figs. 114–1 and 114–2). This surface-dependent reaction is enhanced by the EPCR that binds protein C.6,9,10 With the aid of its nonenzymatic cofactor, protein S, as well as other potential lipid and protein cofactors, APC inactivates factors Va and VIIIa by highly selective proteolysis, yielding inactive (i) cofactors, that is, factors Vi and VIIIi (see Fig. 114–3 and Chap. 113, Figs. 113–11 and 113–13). Protein S also can directly inhibit factors VIIIa, Xa, and Va.11,12,13 Thus, APC and protein S inhibit multiple steps in the intrinsic coagulation pathway.
At each step in the coagulation pathways, each clotting protease can be inhibited by one or more plasma protease inhibitors in reactions stimulated by negatively charged glycosaminoglycans such as heparan sulfate or heparin (see “Inhibition of Coagulation Proteases by Protease Inhibitors” below). Given the highly nonlinear nature of the coagulation pathways with both positive and negative feedback reactions, synergy between the protein C pathway and plasma protease inhibitors is important for regulating thrombin generation.
There is continuous activation of coagulation factors at a basal physiologic low level. Plasma from all normal subjects contains circulating active enzymes, factor VIIa,14 and APC,15 as well as various polypeptide fragments generated by the action of clotting proteases, namely fibrinopeptides,16,17 prothrombin fragment 1+2,18 and activation peptides for factors IX and X.19,20 The presence of multiple clotting factors that require positive feedback activation (e.g., factors V, VIII, XI, and VII) imparts special threshold properties to the blood coagulation pathways, making the coagulant response nonlinearly responsive to stimuli. Theoretical analysis of blood coagulation as a threshold system suggests there can be an all-or-none response to various levels of stimulation, depending on the ensemble of activating and inhibitory reactions that defines upregulation and downregulation of thrombin generation.21,22 The coagulation system is active, but idling, and is poised for extensive and explosive generation of thrombin. Because of synergy among various cellular and humoral anticoagulant mechanisms that establish a threshold system, the presence of multiple coagulation inhibitors with complementary modes of action prevents massive thrombin generation in the absence of a substantial procoagulant stimulus.
HEREDITARY DEFICIENCIES ASSOCIATED WITH THROMBOTIC DISEASE
Evidence for the physiologic importance of specific factors for controlling coagulation reactions comes from clinical observations and animal model studies. Major identified genetic risk factors for venous thrombosis involve protein structural defects in factor V, protein C, protein S, and antithrombin (Chap. 130). There are also gene regulatory defects associated with thrombotic disease, such as the G20210A polymorphism in the prothrombin gene that causes elevated levels of prothrombin, and defects in protein C gene regulatory elements that decrease the expression of protein C. Deficiencies of thrombomodulin might also be associated with increased risk of arterial thrombosis. Association of hereditary abnormalities of EPCR with increased risks of thrombosis has been suggested, but this remains somewhat controversial.
PROTEIN C PATHWAY COMPONENTS
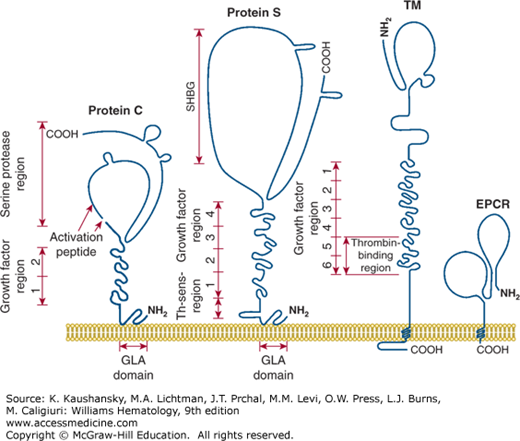
Figure 114–5 is a schematic of the structures of protein C, protein S, thrombomodulin, and EPCR. These proteins contain multiple domains, each of which may mediate different molecular functions. Values for the molecular weight, normal plasma concentration, chromosomal location, and gene structures of these factors are given in Table 114–1. Factors Va and VIIIa, as substrates of APC, are also participants in the reactions of the anticoagulant protein C pathway. Moreover, factor V, but not factor V Leiden, appears to act as an APC cofactor for the inactivation of factor VIIIa (see “Factor V as Activated Protein C Cofactor” below).23
Figure 114–5.
Membrane-bound protein C, protein S, thrombomodulin (TM) and endothelial cell protein C receptor (EPCR). Each protein is a multidomain protein that extends above the surface of cell membranes, and different domains mediate different functions of each protein. Protein C and protein S can bind reversibly to phospholipid membranes through their NH2-terminal γ-carboxyglutamic acid (GLA) domains which contain nine or 11 GLA residues that bind four to six Ca2+ ions. TM and EPCR are integral membrane proteins that are embedded in cell membranes by a single hydrophobic transmembrane sequence. (Adapted with permission from Esmon CT: The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem 264(9):4743–4746, 1989.)
| Molecular Weight (kDa) | Plasma Concentration (mcg/mL) | Half-Life (h) | Chromosome | Gene (kb) | Exon (N) | Function | |
|---|---|---|---|---|---|---|---|
| Protein C | 62 | 4 | 6 | 2q13–14 | 11 | 9 | Anticoagulant protease |
| Protein S | 75 | 26 | 42 | 3p11.1–11.2 | 80 | 15 | Activated protein C (APC)-cofactor and coagulation inhibitor |
| Thrombomodulin | 60–105 | 0.020 | ND | 20p11.2–cen | 3.7 | 1 | Receptor for thrombin/protein C |
| Endothelial protein C receptor (EPCR) | 46 | 0.098 | ND | 20q11.2 | 6 | 4 | Receptor for protein C/APC |
| Protease-activated receptor-1 (PAR-1) | 68 | NA | NA | 5q13 | 27 | 2 | G-protein–coupled receptor |
| Antithrombin | 58 | 150 | 70 | 1q23–25 | 14 | 7 | Protease inhibitor |
| Tissue factor pathway inhibitor (TFPI) | 34 | 0.1 | ND | 2q31–32.1 | 85 | 9 | Protease inhibitor |
| Heparin cofactor II | 66 | 70 | 60 | 22q11 | 16 | 5 | Protease inhibitor |
| Protein Z | 70 | 1.7 | 60 | 13q34 | 9 | Plasma protein | |
| Protein Z–dependent protease inhibitor (ZPI) | 72 | 1.5 | 14q32.13 | 5 | Protease inhibitor |
In 1976, Stenflo designated a bovine plasma vitamin K–dependent protein that eluted in the third peak (peak C) from an anion exchange column as bovine “protein C.”24 Protein C, actually previously described as the anticoagulant factor autoprothrombin II-A,25 is a plasma serine protease zymogen that can be converted to an active serine protease by the action of thrombin.
Protein C is synthesized in the liver as a polypeptide precursor of 461 residues, with a prepropeptide of 42 amino acids that contains the signal for carboxylation of Glu residues by a carboxylase that forms nine γ-carboxyglutamic acid (GLA) residues and secretion of the mature protein.26,27,28 The mature glycoprotein of Mr 62,000 contains 419 residues (see Chap. 113, Fig. 113–1 and Fig. 114–5) and N-linked carbohydrate, and the majority of the secreted protein C molecules are cleaved by a furin-like endoprotease that releases Lys156-Arg157 and generates a two-chain zymogen that circulates in plasma at 65 nM (4 mcg/mL).29 The heavy and light chains of plasma protein C are covalently linked by a disulfide bond that keeps the serine protease globular domain (residues 170 to 419) covalently tethered to the N-terminal string of three domains, the GLA domain and the epidermal growth factor (EGF)-like domains EGF1 and EGF2.26,27,28,29,30
The GLA domain of protein C (residues 1 to 42) and APC is important for a number of functions, including binding to phospholipid-containing membranes (see Chap. 113, Fig. 113–3), thrombomodulin, and EPCR; thus, incomplete carboxylation impairs the functional anticoagulant activity of APC.31,32,33 The two EGF modules in the light chain may contribute to interactions of APC with protein S and of protein C with thrombomodulin.
The serine protease domain of protein C is homologous to other trypsin-like proteases, and three-dimensional modeling34 and X-ray crystallographic structures30 reflect the structural similarity of APC to members of the serine protease family of which chymotrypsin is the prototype (see Mather and colleagues30 for conversion of protein C to chymotrypsin numbering). APC’s trypsin-like protease domain exerts its anticoagulant activity by highly specific interactions with factors Va and VIIIa followed by cleavage at only two Arg-containing peptide bonds in factors Va and VIIIa (see “Factors Va and VIIIa as Substrates for Activated Protein C” below). These stereo-specific interactions involve both the APC enzymatic active site region and a number of APC residues that are termed exosites because they are not located in the immediate vicinity of APC’s enzymatic active site. Such APC exosites are essential for specific recognition of the macromolecular substrates, factors Va and VIIIa as well as recognition of cellular APC receptors.35–44
Purified plasma protein C concentrate (Ceprotin) is FDA-approved for treating protein C–deficient patients.45 Recombinant APC (Xigris) reduced all-cause 28-day mortality in severe sepsis adult patients in the PROWESS phase III trial in 2001 and was FDA-approved for this indication.46 This successful therapy of adult severe sepsis using APC followed preclinical antithrombotic and sepsis studies in baboons.47,48 However, a decade later, in the PROWESS-SHOCK trial, recombinant APC did not reduce mortality in adult severe sepsis patients,49,50,51 and Xigris was withdrawn from the market. Animal injury model studies have also shed light on in vivo mechanisms for APC beneficial effects in many preclinical injury models and in many models the pharmacologic benefits of APC appear to be independent of APC’s anticoagulant actions (see “Activated Protein C Direct Cellular Activities” below).7,8
The protein C gene, comprising nine exons and eight introns, is located on chromosome 2q14–21 and spans 11 kb (see Chap. 113, Fig. 113–10, and Table 114–1).52 The protein C gene is homologous to the genes for factors VII, IX, and X (Chap. 113).
Hereditary protein C deficiency associated with thrombosis is caused by numerous mutations (see protein C mutation databases).53,54 Based on three-dimensional structures of the protein C, the structural basis for hereditary protein C defects has been rationalized.34,55,56 Most mutations that cause type I protein C deficiency, characterized by parallel reductions in activity and antigen, involve amino acid residues that form the hydrophobic cores of the two folded globulin-like domains that are characteristic of serine proteases. These mutations destabilize either the process or the product of protein folding, and they result in unstable molecules that are poorly secreted and/or exhibit a very short circulatory half-life. In contrast, most mutations that cause type II defects (reduced anticoagulant or enzymatic activity but normal antigen levels), that is, circulating dysfunctional molecules, involve polar surface residues that do not affect polypeptide folding or thermodynamic stability; these polar residues presumably are involved in protein–protein interactions important for expression of anticoagulant activity.
Rare variants derived from a founder’s mutation appear to be distinctive for races. Two protein C mutations, Arg147Trp and a Lys150 deletion, are significant venous thrombosis risk factors in Chinese but not in Americans of European descent or Japanese.57,58,59
Severe protein C deficiency as a consequence of homozygous knockout of the mouse protein C gene showed a similar phenotype as severe human protein C deficiency (Chap. 130), with perinatal consumptive coagulopathy in the brain and liver and either death or massive thrombosis that occurred either intrauterine or shortly after birth.60
Plasma “protein S,” which was named in honor of Seattle, the city of its discovery, is a vitamin K–dependent glycoprotein61 that is synthesized by hepatocytes, neuroblastoma cells, kidney cells, testis, megakaryocytes, and endothelial cells, and is also found in platelet α-granules.62
Protein S is synthesized as a precursor protein of 676 amino acids, which gives rise to a mature secreted single-chain glycoprotein of 635 residues with three N-linked carbohydrate side chains (see Fig. 114–5).63,64 Eleven GLA residues in the N-terminal region of mature protein S contribute to Ca2+-mediated binding of the protein to phospholipid membranes. The thrombin-sensitive region, residues 47 to 72, follows the GLA-domain (see Fig. 114–5).
The C-terminal region of protein S, residues 270 to 635, the sex hormone–binding globulin-like (SHBG) region contains binding sites for C4b-binding protein (see “Activated Protein C–Independent Anticoagulant Activity of Protein S” below)65 and for factor V, as well as factor Va.66,67 Protein S, like the homologous gas6, also binds to receptor tyrosine kinases, for example, Axl, and initiates cell signaling, and the SHBG region binds the receptor.68 Thus, for the expression of its multiple activities, different domains of protein S exhibit a number of different binding sites for different proteins.
The protein S gene, comprising 15 exons and 14 introns, is located on chromosome 3p11.1–11.2 and spans 80 kb (see Fig. 113–16 and Table 114–1).69,70 The protein S gene has limited homology with other genes for vitamin K–dependent factors in the GLA and EGF domains and notable homology of the region coding for residues 240 to 635 with genes of the SHBG family. Humans contain a protein S pseudogene that contains several stop codons and is not translated and that is located very near the normal protein S gene on chromosome 3.
The molecular basis for hereditary protein S deficiency associated with venous thrombosis (Chap. 130) is linked to more than 100 different mutations.71 A protein S polymorphism that is strongly linked to risk for venous thrombosis in Japanese subjects is known as protein S Tokushima. It involves K155E, which ablates APC-cofactor activity.57,72 But the K155E apparently is not present in Americans of European ancestry or Chinese populations, thus mirroring the presence of factor V Leiden and prothrombin G20210A that are risk factors in Americans of European decent but not in the Japanese, Chinese, or Americans of African descent populations.73 Another single nucleotide polymorphism present in approximately 1 percent of Americans of European descent is S460P, which is designated protein S Heerlen; it results in absence of N-linked carbohydrate on Asn458 but has no accepted significant functional consequence.74
Thrombomodulin was discovered as an endothelial cell surface receptor that binds protein C and thrombin, thereby accelerating protein C activation.75,76,77,78 Binding of thrombin to thrombomodulin converts thrombin from a procoagulant enzyme to an anticoagulant enzyme because thrombomodulin-bound thrombin loses its normal ability to clot fibrinogen or activate platelets.79,80 Thrombomodulin is a multidomain transmembrane protein comprising an N-terminal lectin-like domain, six EGF domains, a Ser/Thr-rich region, a single membrane-spanning sequence, and an intracellular C-terminal tail (see Fig. 114–5).5,6,7,8,75–83 EGF domains 4, 5, and 6 are essential for activation of protein C, with the latter two domains binding thrombin and the first domain binding protein C. The mature protein has 557-amino-acid residues and variable amounts of N– and O-linked carbohydrate modifications that cause variability in molecular size. Glycosaminoglycans, notably chondroitin sulfate, covalently attached to the Ser/Thr-rich region, contribute to the functional properties of thrombomodulin by enhancing either protein C activation by thrombin or by accelerating neutralization of thrombin by protease inhibitors. Modulation of the substrate specificity of thrombin by thrombomodulin involves conformational changes in thrombin caused by binding of thrombomodulin.
Low levels of soluble thrombomodulin circulate in plasma, presumably as a result of limited proteolysis of the protein near its transmembrane cell surface anchor. The functional significance of circulating thrombomodulin is unknown, although variations in its plasma level arise in different clinical conditions.
Recombinant soluble thrombomodulin has been developed for its potential therapeutic value for disseminated intravascular coagulation and has been approved for this indication in Japan.77,84
The thrombomodulin gene, which lacks introns, is located on chromosome 20p11.2 and spans 3.7 kb (see Chap. 113, Fig. 113–19, Fig. 114–5, and Table 114–1).77,82 Deletion of the thrombomodulin gene in mice is embryonically lethal.85 Downregulation of thrombomodulin gene expression is promoted by a variety of inflammatory agents, including endotoxin, interleukin-1, and tumor necrosis factor (TNF)-α, whereas its expression is upregulated by retinoic acid.5,6,7,8,86,87 Generally, thrombomodulin is a key member among the counterbalancing factors that contribute to inflammation, thrombin generation, and coagulation in the endothelium.
Thrombomodulin mutations are well documented in atypical hemolytic uremic syndrome patients,78,88 and they may also be associated with an increased risk of arterial thrombosis and myocardial infarction. In contrast, there is less supportive data for association with risk for venous thrombosis (Chap. 130).89,90,91,92 Atypical hemolytic uremic syndrome is strongly linked to excessive complement activation, and thrombomodulin’s lectin-like domain inhibits complement activation.77,93 Furthermore, besides promoting protein C activation by thrombin, thrombomodulin also supports activation of the carboxypeptidase, also known as thrombin-activatable fibrinolysis inhibitor (TAFI) that is a potent inactivator of bradykinin and of the activated complement components, C3a and C5a.94,95,96
EPCR binds both protein C and APC with similar affinities through their GLA domains and mediates multiple activities of this zymogen or its activated protease, APC.9,10,33,86,97–107 The mature EPCR glycoprotein contains 221-amino-acid residues and N-linked carbohydrate, giving an Mr of 46,000. EPCR is an integral membrane protein that is homologous to CD1/major histocompatibility complex class I molecules. The N-terminus is part of an extracellular domain, which is connected to a single transmembrane sequence that is followed by a short Arg-Arg-Cys-COOH cytoplasmic tail (see Fig. 114–5). The cytoplasmic tail can be palmitoylated, and this modification may help localize EPCR to certain lipid rafts or caveolae. The three-dimensional structure of EPCR determined by X-ray crystallography or inferred by molecular modeling established that the GLA domain of protein C and APC binds to EPCR.107,108 EPCR on endothelial surfaces enhances by greater than fivefold the rate of activation of protein C by thrombin–thrombomodulin (see Fig. 114–2). EPCR is also required for the cytoprotective activities of APC by promoting the cleavage of PAR-1 by APC to induce cell-signaling pathways (see Fig. 114–4). Notably, the cytoprotective actions of APC are completely independent of its anticoagulant activity and are based on cell signaling actions (see “Activated Protein C Direct Cellular Activities” below).7,8,109,110,111
The presence of functional EPCR on the cell surface is regulated by two mechanisms, namely generation of EPCR and clearance of EPCR. Inflammatory mediators induce EPCR ectodomain shedding from the endothelial cell surface by metalloproteinase TNF-α converting enzyme (known as “TACE”).112 The soluble EPCR ectodomain is found in normal human plasma at 100 ng/mL; however, carriers of the H3 EPCR haplotype that includes a Ser219Gly polymorphism (rs867186) have threefold higher soluble EPCR plasma levels.113 Higher plasma levels of soluble EPCR are found in patients with disseminated intravascular coagulation or systemic lupus erythematosus, although plasma EPCR levels are not correlated with pathology-related alterations in circulating thrombomodulin levels.114 Soluble EPCR binds the protein C and APC via their GLA domains with an affinity similar to the membrane-bound receptor. Because binding of the APC GLA domain to negatively charged phospholipid membranes is required for its anticoagulant activity, soluble EPCR at relatively high levels in purified reaction mixtures inhibits the anticoagulant action of APC against factor Va, although it does not block the reaction of APC with protease inhibitors.10,86,99,115
The EPCR crystal structure surprisingly revealed a single phospholipid molecule bound in a surface groove on the protein.107 Secreted phospholipase A2 group V can modify the lipid in EPCR and cause EPCR to lose its ability to bind protein C and APC.116,117 The presence of functional EPCR on the cell surface has important implications for thrombotic and inflammatory vascular disease because EPCR inactivation in vivo increases susceptibility to thrombotic and inflammatory diseases.10,86,97
The physiologic requirement for EPCR in mice was established by the embryonic lethality observed for knockout of the murine EPCR gene.118
EPCR has functionally important interactions with multiple molecules beyond protein C and APC. It binds factor VII and factor VIIa.10 Furthermore, EPCR was recently implicated to play a potentially important role in the pathogenesis of severe malaria.119,120,121,122
The EPCR gene, comprising four exons and three introns, is located on human chromosome 20q11.2 and spans 6 kb (see Table 114–1).123
PAR-1, discovered as a high-affinity human platelet receptor for thrombin,124 is the prototype of a four-member subfamily of G-protein–coupled receptors that share an unusual mechanism of activation, namely activation by proteases.124,125,126,127,128,129,130 Each PAR contains seven transmembrane helical domains and an extracellular N-terminal tail that is cleaved by an activating protease such that the newly generated aminoterminus is a tethered ligand that triggers activation of the coupled G-protein. Human platelets employ PAR-1 and PAR-4 for activation by thrombin whereas, curiously, murine platelets that are devoid of PAR-1 require PAR-3 and PAR-4 for thrombin’s normal effects.125,131 PAR-1 is activated by various plasma proteases132,133,134,135 and is generally required for APC’s cytoprotective activities (see “Cellular Receptors for Physiologic Effects of Activated Protein C on Cells” below).7,8,110,111
The PAR-1 gene contains only two introns, is located on chromosome 5q13, and spans 25 kb (see Table 114–1).125 Much is known about many factors that can either upregulate or downregulate the PAR-1 gene.124,125,126,127,128,129,130
ACTIVATION OF PROTEIN C
Protein C is activated from zymogen to active protease as a result of cleavage by thrombin at the Arg169–Leu170 peptide bond in a reaction that is accelerated by thrombomodulin and EPCR (see Fig. 114–2 and “Thrombomodulin” above).5,7,8,76,81,86 Thrombin infusions into animals generate anticoagulant activity because of APC.136,137 Interestingly, thrombin infusion into hyperlipidemic monkeys with atherosclerosis generates less APC and causes a poorer ex vivo response to APC compared with normolipidemic control monkeys,138 showing that hyperlipidemia and vascular disease can affect protein C activation.
Ischemia causes protein C activation in vivo. A brief occlusion of the left anterior descending coronary artery in pigs results in APC generation.139 During cerebral ischemia in humans undergoing routine endarterectomy, APC increases in the venous cerebral blood.140 Protein C is significantly activated during cardiopulmonary bypass, mainly during the minutes immediately after aortic unclamping in the ischemic vascular beds.141 Streptokinase therapy for acute myocardial infarction increases circulating APC.142
Circulating APC concentration in normal human subjects is highly correlated with circulating levels of protein C zymogen.143 Based on protein C infusion studies in protein C-deficient subjects, the level of circulating APC is strongly determined by the concentration of protein C.144 EPCR appears to be required for normal protein C activation in response to thrombin infusions in experimental animals.145 EPCR and thrombomodulin must be in close proximity on cell surfaces (see Fig. 114–2), although this has yet to be experimentally demonstrated.
Thrombomodulin and EPCR appear to differ markedly in their relative distribution densities on blood vessels as the former is abundantly present in the small blood vessels but less so in large vessels, whereas the latter is more abundant in large vessels than in small vessels.85,86,146,147 Low levels of thrombomodulin are expressed in brain,148 and brain-specific activation of protein C in humans occurs during carotid occlusion.140
Proteolytic cleavage and activation of protein C can also be effected by meizothrombin, plasmin, or factor Xa.149,150,151,152,153 On the surface of cultured endothelial cells, negatively charged sulfated polysaccharides in the presence of phospholipid vesicles containing phosphatidylethanolamine can enhance the rate of protein C activation by factor Xa to approach the protein C activation rate of thrombin: thrombomodulin.152 No data yet indicate whether protein C activation by meizothrombin, plasmin, or factor Xa is physiologically relevant.
Protein C activation is stimulated by platelet factor 4. Both in vitro and in vivo data imply that platelet factor 4 may play a physiologic role in enhancing APC generation and influencing the activities of the protein C system.154,155,156,157
ACTIVATED PROTEIN C ACTIVITIES
The clinical phenotype of severe protein C deficiency in neonatal purpura fulminans implies that APC exerts multiple physiologically essential activities, including potent anticoagulant and antiinflammatory actions (Chap. 130). Recent advances establish that APC’s antiinflammatory actions are but one manifestation of its ability to interact directly with cell receptors to provide multiple cytoprotective activities.7,8,110,111 These two distinct types of activities of APC—intravascular anticoagulant activity and initiation of cell signaling—are mediated by different sets of molecular interactions, and both types of activities are clinically relevant.
Mechanisms for APC’s direct anticoagulant activity involve factors V and VIII, the two homologous coagulation cofactors that circulate as inactive molecules and that are converted to active cofactors by limited proteolysis (see Chap. 113, Figs. 113–11 and 113–13). APC circulates at 40 pM (picomolars) in normal humans, and there is an inverse correlation between fibrinopeptide A, the product that is cleaved from fibrinogen by thrombin, and APC levels in healthy nonsmoking adults, suggesting APC is a significant regulator of basal thrombin activity.15,158
Factors V and VIII are synthesized as large single-chain precursor coagulation cofactors of Mr 330,000, consisting of three homologous A domains (A1, A2, and A3) and two homologous C domains (C1 and C2) with a very large intervening, generally nonhomologous domain, designated the B domain, that connects the A2 and A3 domains (Chap. 113). Activation of the inactive precursor form of the two cofactors V and VIII involves limited proteolysis.23,159,160,161,162,163,164 Factor V activation involves cleavages at Arg709, Arg1018, and Arg1545 by thrombin, factor Xa, or other proteases.23,164,165,166,167,168 Cleavage at Arg1545 is the key step for generating factor Va activity because this proteolysis releases the B domain that blocks binding of factor Xa to factor Va.164,169 The various forms of factor Va (see Chap. 113, Fig. 113–11) are composed of two polypeptide chains, one bearing the A1-A2 domains and the other bearing the A3-C1-C2 domains. Although generally similar to factor V activation, factor VIII activation (see Fig. 113–13) involves formation of a heterotrimer of polypeptide chains containing the A1 domain, the A2 domain, and the A3-C1-C2 domains, respectively. In contrast to heterodimeric factor Va, heterotrimeric factor VIIIa is intrinsically unstable as a consequence of spontaneous dissociation of the A2 domain.170
Irreversible proteolytic inactivation of factors Va and VIIIa by APC can be accomplished by proteolysis at Arg506 and Arg306 in factor Va and Arg562 and Arg336 in factor VIIIa (see Chap. 113, Figs. 113–11 and 113–13).23,171,172,173 Currently, the most common identifiable venous thrombosis risk factor involves a mutation of Arg506 to Gln in factor V that results in APC resistance (Chap. 130). The complexities of APC-dependent inactivation of factor Va and VIIIa are compounded by the number of different molecular forms of Va and VIIIa that can be generated by limited proteolysis by a variety of proteases and by their differing susceptibilities to APC and to the different APC cofactors.
APC resistance is defined as an abnormally reduced anticoagulant response of a plasma sample to APC (Chap. 130) and can be caused by many potential abnormalities in the protein C anticoagulant pathway. Such abnormalities could include defective APC cofactors, defective APC substrates, or other molecules that interfere with the normal functioning of the protein C anticoagulant pathway (e.g., autoantibodies against APC, APC cofactors, or APC substrates).
A report of familial venous thrombosis associated with APC resistance without any identifiable defect in four Swedish families174 led to an intensive search for a genetic explanation that was soon found to involve replacement of G by A at nucleotide 1691 in exon 10 of the factor V gene which causes the amino acid replacement of Arg506 by Gln.175,176,177 This factor V variant, like the prothrombin variant nt G20210A, arose in a single white founder some 18,000 to 29,000 years ago178,179 and is known as Gln506-factor V or factor V Leiden. This mutation is currently a common, but not the only, cause of APC resistance (Chap. 130).
The molecular mechanism for APC resistance of Gln506-factor V is based on the fact that the variant molecule is inactivated 10 times slower than normal Arg506-factor Va.23,177,180,181,182 The variant factor Va exhibits only a partial resistance to APC because cleavage at Arg306 in factor Va also occurs, causing complete loss of factor Va activity.
Plasma and recombinant factor V can exist in two biochemically distinct forms, designated factor V1 and factor V2 that differ in N-linked carbohydrate on Asn2181, near the phospholipid binding region of the C2 domain as factor V2 has none.183,184 Because the N-linked carbohydrate appears to decrease the apparent affinity of factor V1 or Va1 for phospholipid, it reduces the specific clotting activity and susceptibility to APC. Normal plasma contains a mixture of factors V1 and V2. Removal of the carbohydrate attached to factor V increases the rate of inactivation of factor Va by APC, although the clinical significance of this phenomenon is unknown.185
APC resistance with no identifiable genetic or acquired abnormalities is well described in patients with venous and arterial thrombosis and, at least for research purposes, should be therefore examined in patients with a suspected thrombophilia. Further studies are needed to identify the causes of APC resistance in such patients.186,187,188 One major challenge involves defining the normal range for the clotting assays that are actually used to characterize APC resistance and the multiple plasma analytes or nonplasma assay components that are present in the assays. For example, activated partial thromboplastin time-based assays are not equivalently sensitive as are dilute tissue-factor-based assays to plasma high-density lipoprotein (HDL) levels or oral contraceptive use.189,190,191 Plasma variables, such as elevated prothrombin levels,192,193 may affect the response to APC by inhibiting APC anticoagulant actions. Endogenous thrombin potential assays involving dilute tissue factor as the procoagulant initiator provide additional tools for defining and characterizing APC resistance and extend the tools for shedding light on the gray area of APC resistance found in some thrombosis patients that is not linked to currently known factors.
APC anticoagulant activity is enhanced by a number of factors that may be termed APC anticoagulant cofactors; these include Ca2+ ions; certain, but not all, phospholipids; protein S; factor V; certain glycosphingolipids; and HDL.
Certain phospholipids, such as phosphatidylserine, phosphatidylethanolamine, and cardiolipin, enhance the anticoagulant activity of APC. In addition, phosphatidylethanolamine and cardiolipin stimulate the APC anticoagulant pathway activities much more than they stimulate the procoagulant pathway activities.194,195,196,197
Protein S structure–activity relationships are informed by much biochemical work and the large number of mutations.71,198 Protein S, as an anticoagulant APC cofactor, forms a 1:1 complex with APC and enhances by 10- to 20-fold the rate of APC’s cleavage at Arg306 in factor Va but not the Arg506 cleavage.181,182 Part of the mechanism for this activity of protein S may be related to its ability to bring the active site of APC closer to the plane of the phospholipid membrane on which the APC–protein S complex is located when the complex is formed.199,200 Protein S also facilitates the action of APC against factor VIIIa.201 Protein S enhances APC’s action, in part at least, by ablating the ability of factor Xa to protect factor Va from APC.202 The GLA domain, thrombin-sensitive region, and EGF1 and EGF2 domains of protein S are implicated in binding APC for expression of anticoagulant activity by the APC–protein S complex.198,203,204,205,206 Cleavage of the thrombin-sensitive region by thrombin abolishes normal binding of protein S to phospholipid and its normal APC-cofactor anticoagulant activity.205,207
Factor V apparently can have anticoagulant as well as procoagulant properties because it enhances the anticoagulant action of APC against factors VIIIa and Va in a reaction in which protein S acts synergistically with factor V.23,208,209,210,211 Cleavage at Arg1545, which optimizes factor Va procoagulant activity, ablates the molecule’s anticoagulant cofactor activity. However, when factor V is cleaved at Arg506 by APC, its APC cofactor activity is increased 10-fold. This suggests that Gln506-factor V has two potential prothrombotic defects, namely, resistance of the variant factor Va to APC inactivation and resistance of the variant factor V to activation of its APC cofactor function.23,209,210,211




