Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .


Introduction
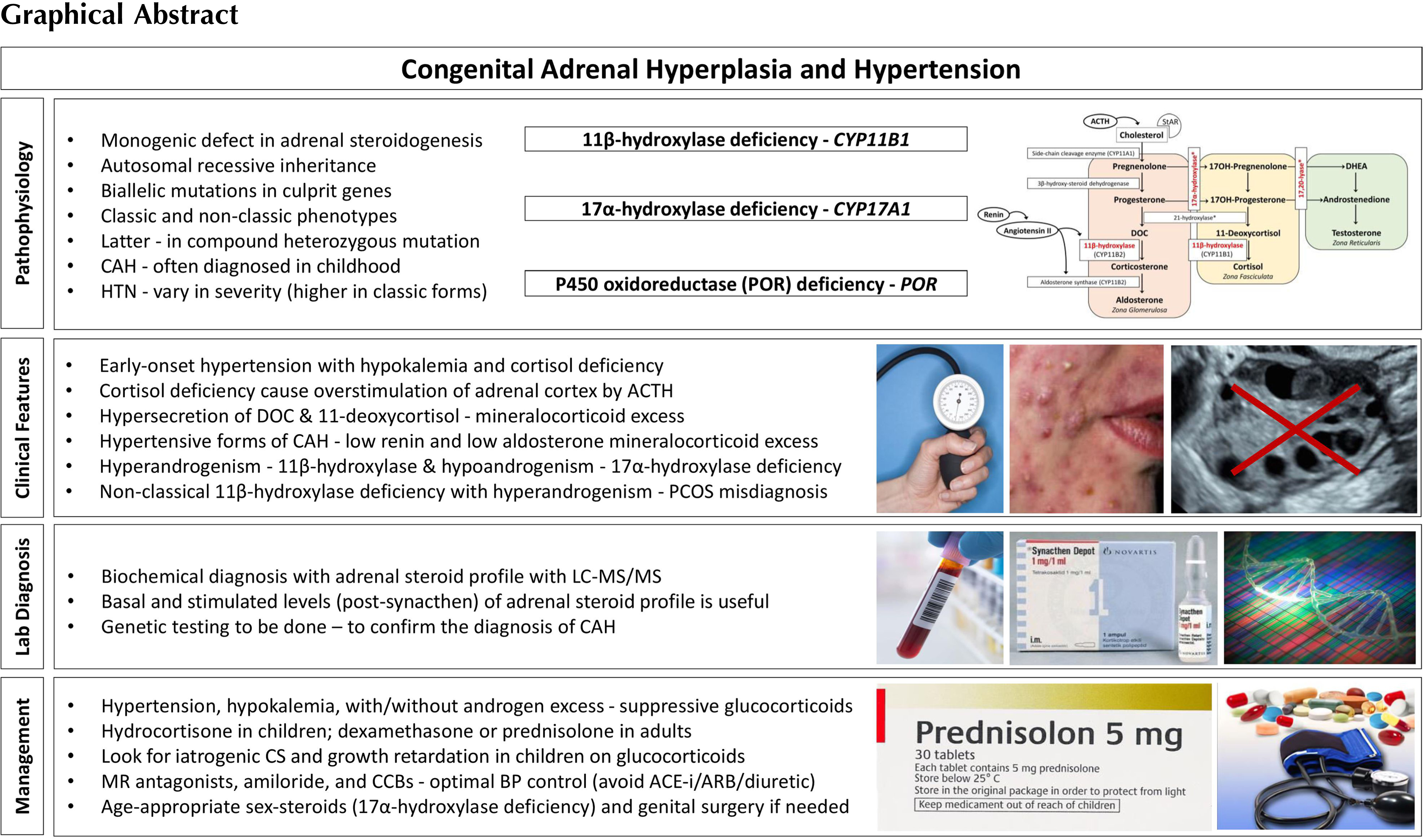
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders caused by variations in the genes encoding enzymes and cofactor proteins which are involved in the adrenal steroidogenesis [ ]. These disorders are often diagnosed in childhood. CAH due to 21-hydroxylase deficiency, the most common type that constitutes ∼95% of all cases, is not associated with hypertension [ ]. On the other hand, 11β-hydroxylase deficiency (the second common type that constitutes ∼5% of all CAH), and 17α-hydroxylase deficiency (a rare form of CAH) are associated with hypertension due to hypersecretion of DOC (deoxycorticosterone) [ ]. These two disorders are characterized by early-onset hypertension associated with hypokalemia, mediated by hypersecretion of mineralocorticoids. Those with partial enzyme defects can present with hypertension in adult life. The mineralocorticoid excess state is associated with a reduction in the renin and aldosterone secretion. Cytochrome P450 oxidoreductase (POR) deficiency is a rare form of CAH, which may also present with hypertension, similar to 17α-hydroxylase deficiency [ , ]. Other rare forms of CAH that are not associated with hypertension and hypokalemia include 3β-hydroxysteroid dehydrogenase type 2 deficiency, congenital lipoid adrenal hyperplasia (due to a defect in the transport protein namely steroidogenic acute regulatory protein or StAR), and cholesterol side-chain cleavage enzyme deficiency [ ]. The CAH subtypes, in the order in which they are mentioned above, are associated with mutations in the following genes: CYP21A2, CYP11B1, CYP17A1, POR, HSD3B2, StAR, and CYP11A1 , respectively. The incidence of CAH due to 21-hydroxylase deficiency is ∼ 1 in 10,000; that due to 11β-hydroxylase deficiency is ∼ 1 in 100,000; and that due to 17α-hydroxylase deficiency is ∼ 1 in 1,000,000 [ ].
Genetic defects in CAH reduce the activity of the enzymes necessary for cortisol production, leading to chronic overstimulation of the adrenal cortex. Thus, except for StAR and cholesterol side-chain cleavage enzyme deficiency, cases with CAH show the clinical features of both deficient steroids distal to and excessive steroids proximal to the enzyme block [ ]. Impaired enzyme function at each step of adrenal steroidogenesis creates a unique steroid profile as a combination of
Genetics of CAH (in general—all forms)
All forms of CAH are monogenic with autosomal recessive inheritance pattern. Most patients have compound heterozygote mutations. The clinical presentation is consistent with the allele that results in a more functional enzyme, and there is usually a genotype-phenotype correlation [ ]. The genetic studies of CAH have provided insight into the pathophysiology and subtle clinical aspects of the disease and may provide prognostic information on disease severity. Furthermore, genotyping is essential in confirming carrier status and is useful for genetic counseling because biochemical methods typically cannot distinguish heterozygotes from the normal population, and assess the disease severity [ ]. The genes for different variants of CAH are well characterized, and the mutation analysis is widely available. Genetic causes of CAH are illustrated in Table 9.1 .
| Gene (OMIM number) | Chromosome | Affected organs | Hypertension | |
|---|---|---|---|---|
| 21-hydroxylase deficiency | CYP21A2 (#201910) | 6p21.1 | Adrenal glands | No |
| 11β-hydroxylase deficiency | CYP11B1 (#202010) | 8q21 | Adrenal glands | Yes |
| 17α-hydroxylase/17,20 lyase deficiency | CYP17A1 (#202110) | 10q24.3 | Adrenal glands and gonads | Yes |
| 3β-hydroxysteroid dehydrogenase type 2 deficiency | HSD3B2 (#201810) | 1p13.1 | Adrenal glands and gonads | No |
| P450 oxidoreductase deficiency | POR (#201750) | 7q11.2 | Adrenal glands, gonads, liver, skeletal structures | Yes |
| Lipoid adrenal hyperplasia | StAR (#600617) | 8p11.2 | Adrenal glands and gonads | No |
| Cholesterol side-chain cleavage enzyme deficiency | CYP11A1 (#118485) | 15q23-q24 | Adrenal glands and gonads | No |
Although biochemical differential diagnosis and genetic diagnosis in rare forms of CAH are not compelling, the diagnosis of 21-hydroxylase deficiency, the most common type can be complicated due to different and misleading clinical presentation, relevant gene region, and mutations. 21-hydroxylase deficiency is caused by inactivating biallelic mutations in the CYP21A2 gene that encodes the microsomal P450 enzyme, 21-hydroxylase ( CYP21A2 , P450c21). The CYP21A2 are located on chromosome 6p21.3, approximately 30 kilobases (kb) apart from the nonfunctional CYP21A1P pseudogene. CYP21A2 and CYP21A1P both consist of 10 exons and share high nucleotide homology of about 98% and 96% in exons and introns, respectively [ , ]. To date, more than 300 mutations in the CYP21A2 gene have been identified. Over 90% of mutations causing 21-hydroxylase deficiency are the result of intergenic recombinations in the HLA region within the 30 kb tandem repeat [ , , ]. Most of the studies conducted to date show that there is a genotype–phenotype relationship, especially in severe salt-wasting and mild nonclassic forms [ , , ].
The 11β-hydroxylase enzyme has two isoenzymes encoded by the CYP11B1 and CYP11B2 genes on chromosome 8q21-q22. Both are located in the inner membrane of mitochondria. CYP11B1 gene mutations result in 11β-hydroxylase deficiency and expression of 11β-hydroxylase enzyme is induced by adrenocorticotropic hormone (ACTH) in the zona fasciculata and zona glomerulosa, whereas CYP11B2 is primarily expressed in zona glomerulosa and is controlled mainly by angiotensin II and potassium levels. Biallelic mutations of CYP11B1 cause 11β-hydroxylase deficiency and more than 100 mutations have been identified [ ]). Mutations with a founder effect in the CYP11B1 gene have been described in some ethnic groups, for example, the p.Q356X and p.G379V mutations in Tunisians, the p.R448H mutation in Moroccan Jews, the p.N394Rfs∗37 and p.L299P mutations in Turks [ , ]. The genotype–phenotype relationship has been reported in many studies [ , ]. As in 21-hydroxylase deficiency, 11β-hydroxylase deficiency may present as classic or nonclassic forms depending on the degree of clinical severity and the percentage of enzyme activity. In the literature, less than 20 mutations associated with the nonclassic form have been described [ , , ]. Milder clinical presentation was observed in compound heterozygous mutations when one of the two alleles was milder than when both were severe, as in 21 hydroxylase deficiency [ ].
Biallelic mutations of CYP17A1 encoding 17α-hydroxylase and 17,20-lyase cause a lack of 17α-hydroxylation in progesterone and pregnenolone, followed by impaired formation of the sex steroids, dehydroepiandrosterone (DHEA), and androstenedione [ ]. CYP17A1 is located on chromosome 10q24.3 and more than 100 mutations have been described to date. Since this gene is expressed in both the adrenal and gonads, its mutations also result in a deficiency in sex steroids which causes sexual infantilism and puberty failure. The majority of identified mutations are associated with the classical phenotype of combined 17α-hydroxylase/17,20-lyase deficiency. Fewer CYP17A1 missense variants have been reported to exhibit partial impairment of 17α-hydroxylase/17,20-lyase activity in 46,XY individuals with mild or absent hypertension and ambiguous genitalia [ ]. Hypertension was not observed in a small number of individuals defined as isolated 17,20-lyase deficiency [ ].
Cytochrome P450 oxidoreductase enzyme deficiency, caused by mutations in the POR gene, was first described in 2004 [ ]. More than 130 cases have been documented. The clinical phenotype in POR deficiency depends on how much and which enzyme function the POR mutation affects [ ]. A287P is the predominant mutation in patients of European ancestry that severely disrupts the 17α-hydroxylase activity and presents with hypertension [ ].
Pathophysiology of hypertension in CAH
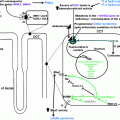
The adrenal cortex has three embryonic regions: The zona glomerulosa synthesizes aldosterone, the most important mineralocorticoid which regulates electrolyte excretion and intravascular volume mainly by its effects on the renal distal tubules and cortical collecting ducts. The zona reticularis and zona fasciculata share the same enzyme pathways to produce cortisol and weak androgens, DHEA, and androstenedione under the control of hypothalamic–pituitary–adrenal axis through ACTH. During the production of cortisol, there are several intermediate precursors, especially DOC and 11-deoxycortisol, that have mineralocorticoid effects [ , , ]. Simplified adrenal steroidogenesis pathway is shown in Fig. 9.1 .
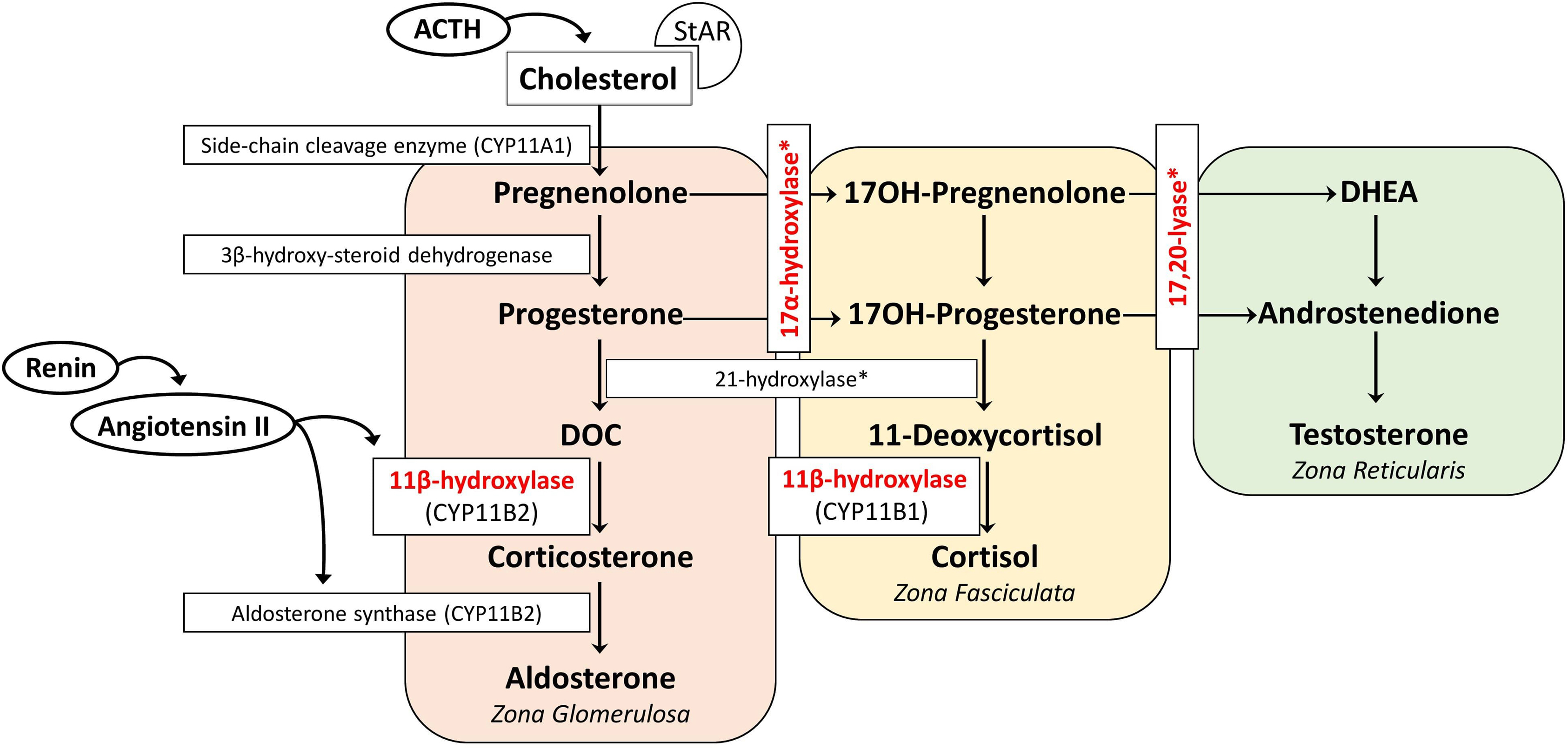
Aldosterone is primarily regulated by the renin–angiotensin system. Hyperkalemia and ACTH are less effective on aldosterone release. Angiotensin II is the major stimulant for the release of aldosterone, and as a potent vasoconstrictor, its level increase when there is a decrease in effective blood volume. Aldosterone exerts its effect by increasing the sodium reabsorption and potassium excretion by the kidney [ ]. Consequently, the net effect is to increase the plasma and extracellular volume and to reduce the renin and aldosterone secretion [ ]. Excessive secretion of aldosterone or other mineralocorticoids suppresses the renin–angiotensin system, resulting in extracellular fluid volume expansion, hypertension, and consequently low renin and low aldosterone concentrations, hypokalemia, metabolic alkalosis (by the feedback mechanism), but the ability to produce aldosterone remains [ , ]. Renin–angiotensin system suppression may not occur in the neonatal period due to renal mineralocorticoid resistance present in the first few months of life [ ]. Of the CAH subtypes, 11β-hydroxylase, 17α-hydroxylase/17,20-lyase, and POR deficiency are associated with hypertension due to the excessive production of DOC.
11β-hydroxylase converts DOC to corticosterone in the zona glomerulosa and 11-deoxycortisol to cortisol in the zona fasciculata. Patients with 11β-hydroxylase deficiency lack the cortisol production, leading to chronic overstimulation of the adrenal cortex by ACTH and accumulation of precursors with mainly mineralocorticoid effects ( Fig. 9.2 ). However, some authors suggest a role for 18-hydroxy and 19-nor metabolites of DOC to have a stronger mineralocorticoid effect and are the main causes of hypertension in 11β-hydroxylase deficiency [ , ]. This hypothesis was established based on the facts that DOC having only weak mineralocorticoid activity when administered to humans or other animals, and DOC levels and blood pressure are not well correlated [ , ]. Hypokalemia, which is a result of excessive mineralocorticoid activity, is also not correlated with hypertension [ ] although these steroids have not been documented to be elevated in 11β-hydroxylase deficiency, presumably requiring hydroxylations mediated mainly by 11β-hydroxylase during their synthesis [ ]. Glucocorticoid treatment can normalize DOC and renin levels leading to reduction of excessive mineralocorticoid activity, but control of hypertension may require additional antihypertensive drugs [ , , , ].
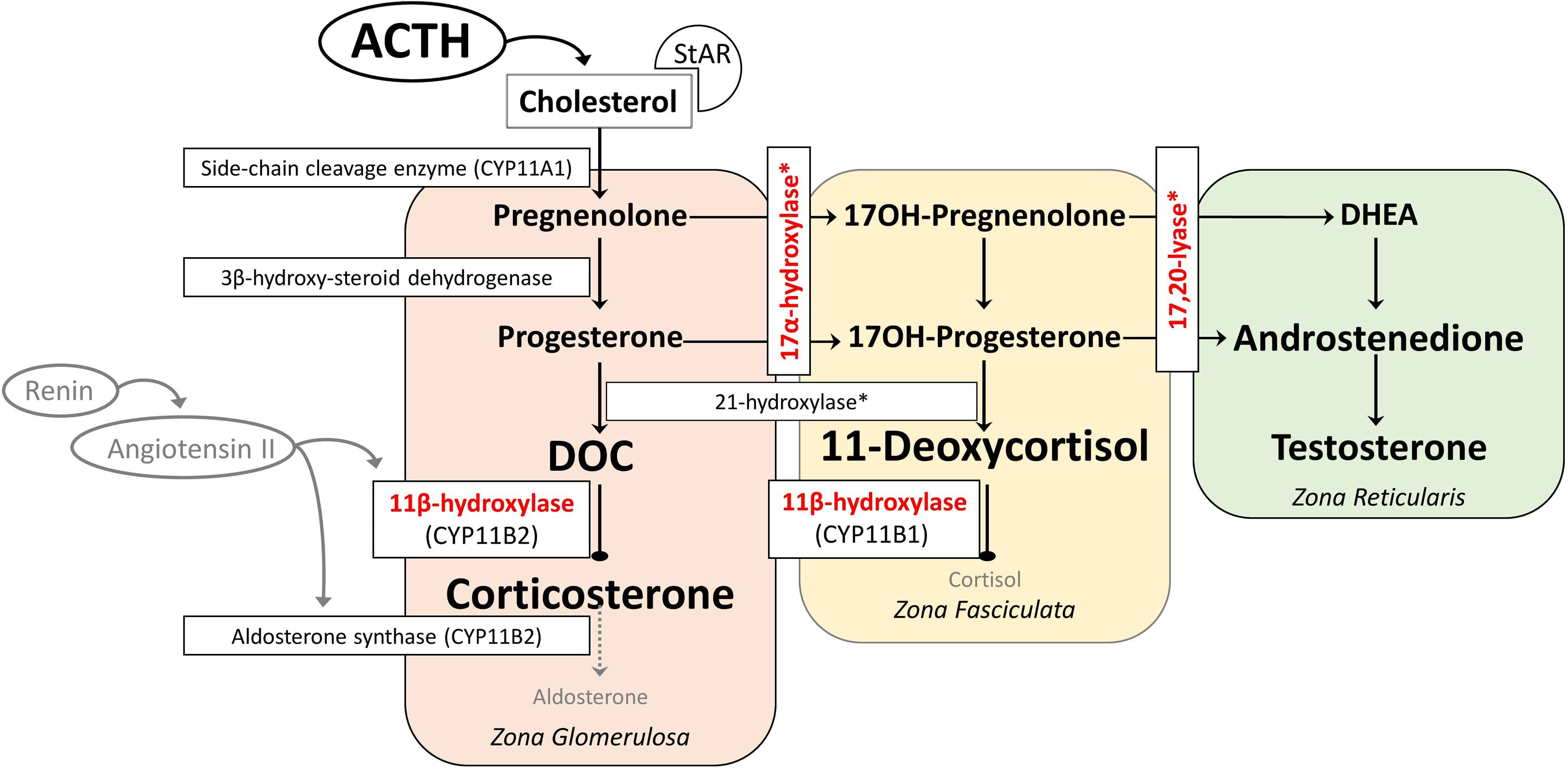
17α-hydroxylase converts pregnenolone to 17-hydroxypregnenolone and progesterone to 17-hydroxyprogesterone. 17,20-lyase then converts 17-hydroxypregnenolone to DHEA and 17-hydroxyprogesterone to androstenedione, which are the major precursors to the sex steroids (testosterone and estrogen). The lack of cortisol production and therefore chronic overstimulation of the adrenal cortex in 17α-hydroxylase deficiency leads to accumulation of the common steroidogenic precursor progesterone, which then shunts toward the zona glomerulosa pathway to make DOC, corticosterone, and aldosterone ( Fig. 9.3 ). Although these steroids are usually produced in response to renin/angiotensin II in healthy subjects in zona glomerulosa, these steroids are found in excessive amounts in zona fasciculata in 17α-hydroxylase deficiency, and several studies have shown that the production of mineralocorticoids from zona fasciculata is increased by ACTH but not by angiotensin II or postural stimulation [ , ]. Moreover, treatment with glucocorticoids (for ACTH suppression) rapidly reduces DOC and corticosterone levels, confirming the primary effect of ACTH on their formation [ ].
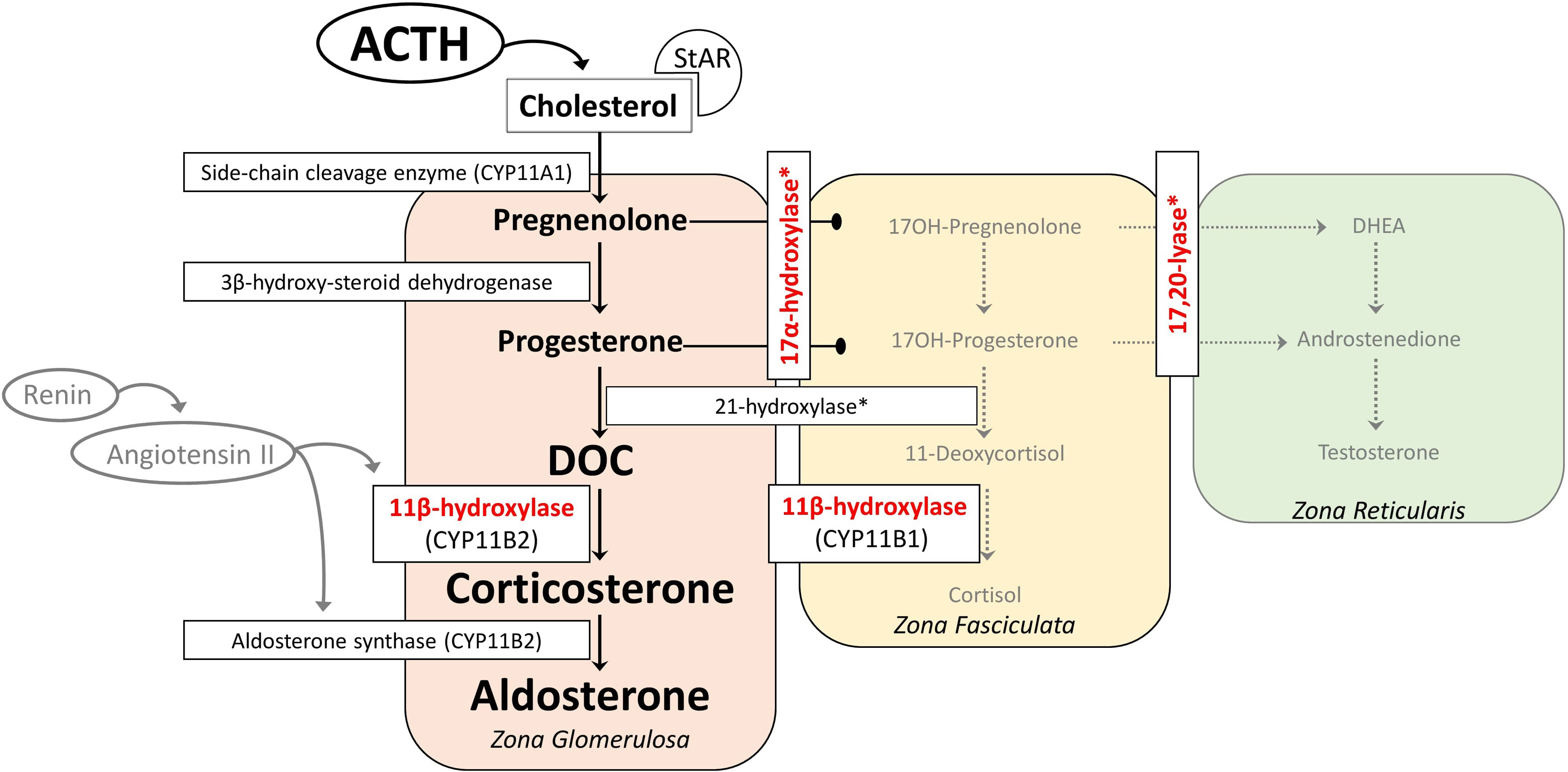
In 17α-hydroxylase deficiency, it seems to be paradoxical that the immediate precursors of aldosterone are elevated, but aldosterone tends to be low. Initially, the second defect in aldosterone synthase was suggested. However, treatment of 17α- hydroxylase deficiency with glucocorticoids allowed aldosterone levels to become normal, indicating an intact aldosterone synthase [ ]. It is assumed that DOC inhibits renin and thus aldosterone synthase, causing sodium retention and volume expansion [ ].
POR plays a role in electron transport in the endoplasmic reticulum, and many enzymes including 17-hydroxylase, 21-hydroxylase and P450 aromatase depend on POR for their catalytic activity. In general, POR deficiency is not manifested with mineralocorticoid deficiency. But mild mineralocorticoid excess can be present and causes arterial hypertension due to particularly 17α-hydroxylase disruption that increases the production of mineralocorticoid intermediates, usually presenting in young adulthood [ , , ].
The mainstay of treatment in classical forms of CAH is glucocorticoid therapy. Hydrocortisone, the most commonly used agent in the treatment, has dose-dependent mineralocorticoid activity. Hypertension as a side effect has been reported with high-dose treatment in patients with CAH [ ].
11β-hydroxylase deficiency
Clinical presentations
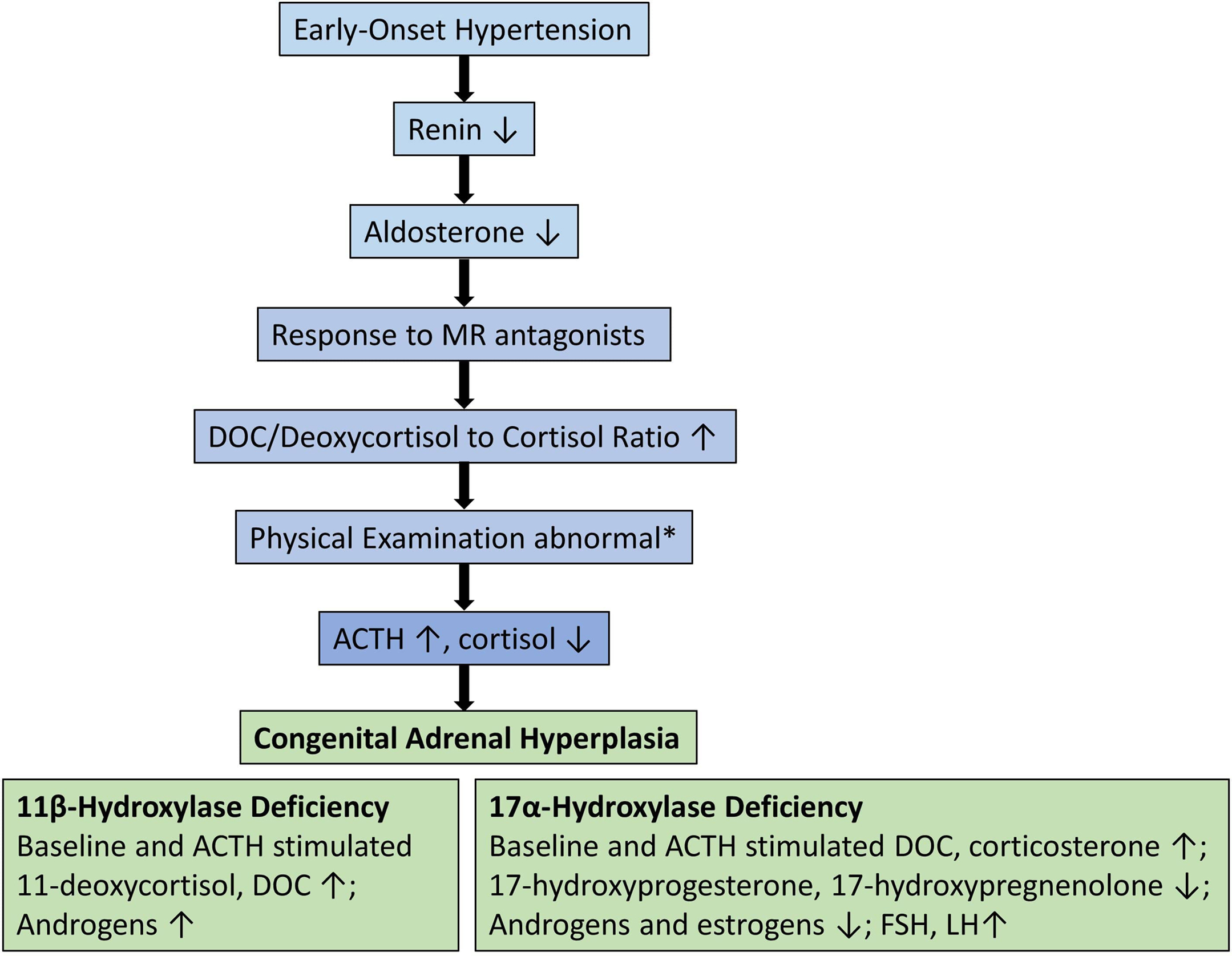
Impaired 11-hydroxylation results in decreased corticosterone and cortisol synthesis, followed by an increase in ACTH, DOC, and 11-deoxycortisol, and the substrate mass effect results in excess production of androgens. This clinically presents as pre- and postnatal virilization of varying severity depending on the degree of enzyme deficiency in both sexes and with hypertension. In the reported series, 66%–80% of girls presented with ambiguous genitalia at birth with enlarged clitoral folds [ , ]. Affected boys appear normal at birth, but premature penile enlargement develops as the first symptom. Patients generally present during infancy and early childhood. Other features include precocious puberty, excessive linear growth, advanced epiphyseal maturation, short adult stature, masculine body habitus, hirsutism, menstrual irregularities, cystic acne, male pattern baldness, increased muscle mass, and hyperpigmentation [ , ]. In the nonclassical form, patients have milder degree of hyperandrogenism and may be misdiagnosed as polycystic ovary syndrome [ , ].
Hypertension of varying severity (mild to moderate) is found in 30%–66% of patients, and is usually diagnosed later in childhood [ , ]. It can be severe and can lead to retinopathy, left ventricular hypertrophy, and cerebrovascular disease [ , ]. Two-thirds of classic 11β-hydroxylase deficiency cases have hypertension at the time of diagnosis, in contrast the ones with nonclassical form are often normotensive at diagnosis [ ].
Diagnostic approach for 11β-hydroxylase deficiency
Initial screening tests in clinically suspected patients include measurement of blood levels of DOC, 11-deoxycortisol, DHEA, androstenedione, and testosterone. Cortisol, renin, aldosterone, and corticosterone are low. Hypokalemia is relatively uncommon and is usually not found in infants. For the given low level of cortisol, ACTH remains elevated [ ]. Although the level of 11-deoxycortisol is very high, it cannot compensate for the lack of cortisol due to low bioactivity. In mild cases (late-onset or nonclassical form) corticotropin-stimulated 11-deoxycortisol and DOC measurements may be helpful (11-deoxycortisol >3 times the upper limit of normal, several times higher for classic) [ ]. Like 21-hydroxylase deficiency, both 3β-hydroxysteroid dehydrogenase type 2 and 11β-hydroxylase deficiencies that are presenting with hyperandrogenemia may have moderate 17OH-progesterone levels. Elevated 17OH-progesterone levels can mislead to the diagnosis of 21-hydroxylase deficiency when used in neonatal screening programs [ ]. Differential diagnosis is made with the 11-deoxycortisol, DOC, and 17-hydroxypregnenolone values [ ]. In recent years, analysis of the adrenal steroid profile with liquid chromatography-tandem mass spectrometry (LC-MS/MS) has been supportive in the differential diagnosis [ , ]. With the neonatal screening programs that include 11-deoxycortisol in the second stage, it is now possible to diagnose and treat cases of 11β-hydroxylase deficiency earlier [ ]. Along with the hormonal evaluation, further genetic testing for the CYP11B1 gene and functional characterization of mutations should be specifically performed to confirm the diagnosis [ ].
Management algorithm
The goals of treatment are to control hypertension, androgen excess, and hypokalemia [ ]. This is achieved by glucocorticoid administration, mostly in the form of hydrocortisone (10–15 mg/m 2 /day in 2–3 divided doses), similar to 21-hydroxylase deficiency [ , ]. In adults, long-acting glucocorticoids such as prednisolone or dexamethasone (2.5–7.5 mg/day and 0.25–0.5 mg/day, respectively) may be preferred [ ]. Glucocorticoid doses should be adjusted to minimize the risk of iatrogenic Cushing’s syndrome [ ]. Stress doses of glucocorticoids are required in acute illness even if the risk of adrenal crisis is less than that in 21-hydroxylase deficiency [ , ].
If the blood pressure remains high despite optimal glucocorticoid therapy, an antihypertensive medication should be added. Spironolactone or amiloride can be used as a single agent or in combination with a calcium channel blocker. Angiotensin-converting enzyme inhibitors and angiotensin receptor II blockers should be avoided, as the renin–angiotensin system is suppressed. Thiazide diuretics should also be avoided or used in combination with a potassium-sparing diuretic, as they may exacerbate or precipitate hypokalemia [ ].
Adult short stature is an important complication on long-term follow-up. If the bone age continues to progress especially in children in their adolescence despite the use of optimal glucocorticoid therapy, aromatase inhibitors are an option [ ]. The peripheral onset of precocious puberty can be centralized, and in this case, gonadotropin-releasing hormone analogue may be started [ ].
Other considerations including individualized treatment plans to modulate patients’ sexual development and surgical correction of genital malformations may be required in females [ ].
Therapeutic targets
The efficacy of treatment is monitored by regular clinical evaluation documenting the degree of virilization, control of hypertension, growth rate, and bone maturation in children. On long-term follow-up, blood pressure should be normalized, normokalemia should be maintained, and DOC levels should decrease even if it is not within the normal range [ , ]. Serum androstenedione and testosterone levels should be evaluated according to age and gender. Higher levels in the postpubertal period are of adrenal origin and indicate inadequate treatment. A measurable renin level indicates adequate suppression of mineralocorticoids but can only occur months after treatment [ ].
17α-hydroxylase deficiency
Clinical presentation
Impaired 17-hydroxylation results in decreased cortisol synthesis, followed by an increase in ACTH, DOC, and corticosterone [ ]. Glucocorticoid deficiency is generally not symptomatic due to the overproduction of corticosterone that has glucocorticoid activity [ ]. In the complete deficiency, due to the deficiency of both 17α-hydroxylase and 17,20-lyase, the adrenal and gonadal sex steroids synthesis is impaired. Patients typically have a female phenotype in most cases, regardless of karyotype and usually present during puberty as girls without secondary sexual characteristics, with hypergonadotropic hypogonadism, and low-renin hypertension [ , ]. Small uterus and ovaries can be detected in 46,XX individuals in childhood, and large cysts in the ovaries due to high gonadotropin levels in adolescence. In partial deficiency, 46,XY individuals usually present with ambiguous genitalia with intraabdominal or inguinal testis during infancy because sufficient androgen cannot be synthesized for external genitalia development [ , ]. Since there is no deficiency of anti-Mullerian Hormone, internal genital development is normal in 46,XY individuals, and Mullerian structures (uterus/cervix) are absent [ , , ]. In the literature, 46,XX individuals misdiagnosed as gonadal dysgenesis and 46,XY individuals misdiagnosed with androgen insensitivity were reported [ , ]. Although patients tend to be taller due to the lack of epiphyseal closure caused by deficient sex steroids, short stature has also been reported in those who are less severely affected with point mutations [ ].
Hypokalemia and hypertension are quite common and usually present from late childhood to adolescence, but their absence does not exclude the disease [ , ]. Even within the same family with the same mutation, varying degrees of hypertension have been reported previously and are thought to be due to factors other than 11β-hydroxylase enzyme activity, such as circulating DOC levels, diet, and environmental factors [ ].
Diagnostic approach
Biochemical diagnosis of 17α-hydroxylase deficiency is established by elevated levels of DOC, corticosterone, ACTH, and gonadotropins with suppressed levels of renin, androgens, and estrogen [ ]. Progesterone is also elevated as in 21-hydroxylase and 11β-hydroxylase deficiency, but 17α-hydroxylase deficiency leads to the absence of sex steroid-dependent puberty development rather than virilization [ ]. Unlike 11β-hydroxylase deficiency, 17-hydroxyprogesterone levels are low and do not rise with the corticotropin stimulation test. Since basal values may be normal in patients with partial deficiency, a corticotropin stimulation test can be performed. The test reveals elevated DOC, pregnenolone, and progesterone [ ]. The progesterone level is high in POR deficiency, but low 17-hydroxyprogesterone and very high DOC distinguish 17α-hydroxylase from POR deficiency. Genetic testing establishes the diagnosis with the identification of the related mutations [ ].
Management algorithm
The goals of the treatment are to prevent glucocorticoid deficiency, to reduce the effects of mineralocorticoid excess and hypertension, and to provide age-appropriate development of secondary sex characters [ ]. When treated with suppressive doses of glucocorticoids, DOC is suppressed and renin and aldosterone levels return to normal range [ ]. The addition of mineralocorticoid receptor antagonists such as spironolactone or eplerenone may be necessary in some cases to treat hypertension. Patients should be treated with age-appropriate sex steroids and genital abnormalities due to virilization should be managed with appropriate surgical procedures [ ].
Therapeutic targets
Blood pressure and serum potassium should be the two main parameters for titration therapy, especially in the first few months. Plasma renin activity can be used as an indicator of mineralocorticoid receptor antagonism but can only be used in the chronic phase as renin may not rise for months or years following chronic suppression [ ].
Overtreatment of CAH
The main treatment option in the classic forms of CAH is chronic glucocorticoid supplements and mostly additional mineralocorticoid therapy. While long-acting glucocorticoid therapy is avoided in children due to its growth-suppressing effects, it is sometimes used in adults. Glucocorticoid therapy aims to prevent adrenal crisis and virilization, allowing nearly normal growth and development during childhood. In general, supraphysiological doses of glucocorticoids are needed to achieve adequate suppression of hormone excesses (10–15 mg/m 2 per day). Management of classic CAH is a difficult balance between hyperandrogenism and hypercortisolism. Laboratory results may be indicative but not always ensure optimal management. Therefore, regular clinical evaluation should always be performed. Overtreatment often leads to Cushingoid side effects such as in linear growth retardation, obesity, osteoporosis, and hypertension [ , , ].
In patients with classic 21-hydroxylase deficiency, mineralocorticoid therapy allows the use of lower doses of glucocorticoids, thus optimizing growth. The only oral mineralocorticoid preparation available is fludrocortisone (9α-fluorocortisol). Hydrocortisone can also activate mineralocorticoid receptors. Approximately 20 mg of hydrocortisone is equivalent to the mineralocorticoid effect of about 0.1 mg of fludrocortisone [ ]. Therefore, when stress doses of hydrocortisone are administered, fludrocortisone treatment is not required [ ].
In infancy, however, mineralocorticoid susceptibility is naturally greater during the first year of life, and they often need higher doses of fludrocortisone therapy than in adults. In this period, finding the appropriate fludrocortisone dose without causing hypertension is the challenge of treatment, especially in the first few years of life, when higher doses and salt supplementation are often needed. In a prospective study, 57.6% of 33 infants with classic CAH, diagnosed by new-born screening, had hypertension in the first 18 months of life [ ].
The majority of studies investigating hypertension in CAH patients, by ambulatory or 24-hour blood pressure measurement, have documented increased systolic and/or diastolic blood pressure values [ , ]. Moreover, even in CAH children without overt hypertension, a reduction in the physiological nocturnal drop response in blood pressure (nocturnal dip) was observed [ , , ].
Subbarayan et al. reported that a large cohort of CAH children, regardless of gender, had significantly higher systolic hypertension (20.9%) compared to the reference population [ ]. However, the prevalence of hypertension was lower than in previous studies, possibly due to the use of lower doses of glucocorticoids and mineralocorticoids in the recent years. Blood pressure was negatively associated with age, possibly due to reduction in fludrocortisone dose [ ].
In a multicenter study, there was a relationship between fludrocortisone dose and blood pressure in 716 patients (2–18 years) diagnosed with 21-hydroxylase deficiency, and hypertension was less common in children who have regular PRA during follow-up. Blood pressure values were significantly correlated to body mass index, age, and the fludrocortisone dose [ ].
Current findings indicate a higher prevalence of hypertension in children and adolescents with classic forms of CAH. In addition to obesity, an overtreatment of glucocorticoids and mineralocorticoids also plays an important pathogenic role.
When to suspect of CAH in patients with hypertension
Clinicians should screen children, adolescents, and young adults who present with hypertension, hypokalemia, and low aldosterone and renin levels. In addition, hyperpigmentation, muscle weakness, delayed or precocious puberty, growth failure, ambiguous genitalia, and virilization are important additional clues. Suspicion for 11β-hydroxylase deficiency should be highest in females with hyperandrogenemia signs such as hirsutism, clitoromegaly, menstrual irregularities, cystic acne, male pattern baldness, increased muscle mass, masculine body habitus, and in males with pseudoprecocious puberty. 17α-hydroxylase deficiency has the highest suspicion in females phenotype without secondary sexual characteristics, with hypergonadotropic hypogonadism. Clinical approaches to CAH-associated hypertension are illustrated in Fig. 9.4 .