Coagulation-Independent Signaling of the Extrinsic Coagulation Pathway
Wolfram Ruf
Tissue and vascular injury requires a rapid hemostatic response that is critically dependent on the tissue factor (TF)-initiated extrinsic coagulation pathway. TF triggers coagulation by forming a catalytic cofactor-enzyme complex with coagulation factor VIIa (VIIa), converting factor X to Xa, and thereby initiating the common downstream coagulation pathway, activating platelets, and converting fibrinogen to fibrin. TF is required for embryonic development in the mouse,1,2,3 and no deficiencies of TF have been reported in humans. The central role of TF-VIIa complex formation for postnatal hemostasis is underscored by the highly similar phenotypes of genetically engineered mice in which TF or VII expression levels are severely reduced. These mice develop cardiac microbleeds, progressive fibrosis, and heart failure.4 Expression of TF in a variety of cell types in mouse and man5,6 supports a role in providing a “hemostatic envelope” around organs and at boundaries to the environment, but TF also has broader functions beyond providing hemostasis and maintaining vascular integrity.
Expression of TF by leukocytes and blood-derived monocytes initiates coagulation that is pivotal for intravascular sequestration of microorganisms in host defense7 and for fibrin formation supporting cell-mediated immune responses in extravascular locations.8,9 Conversely, excessive activation of the TF procoagulant pathways drives disseminated coagulation and lethality in sepsis.10 However, TF is also a direct regulator of myeloid cell-dependent inflammation.11,12 Thus, the concept is emerging that TF-initiated coagulation and direct cell signaling cooperate to protect from infection and regulate innate immune cell function.
TF expression by nonhematopoietic cells also has nonhemostatic functions. TF is synthesized by smooth muscle cells where it can initiate acute intravascular thrombosis following vessel wall injury.13 In addition, TF is upregulated by growth factor stimulation and promotes smooth muscle cell migration and intima hyperplasia.14,15,16,17,18 TF expressed by smooth muscle cells or endothelial cells has been implicated in angiogenic processes by coagulation-independent pathways.19,20,21,22,23 TF is expressed by epithelial cells in the gastrointestinal tract and plays a pivotal role in coagulation activation and inflammatory cell recruitment in experimental colitis.24 TF is distinctly expressed in the basal layers of epithelial barriers in the skin, and reduced wound healing in VII-deficient mice indicates roles for the TF-VIIa complex in stimulating epithelial migration and wound closure.25
The association of TF with cell adhesion integrin receptors,26,27 cytoskeletal structures and cell-cell contacts,28,29,30 and a promigratory G protein-coupled receptor, the protease-activated receptor (PAR)2 provides molecular connections by which TF can support regenerative tissue repair as well as cancer progression. This chapter reviews the basic mechanisms of TF-initiated cell signaling and our current understanding as to how these noncoagulant roles of the TF pathway support physiologic and pathologic processes.
MOLECULAR CELL BIOLOGY OF TF
TF is evolutionary related to the cytokine receptor family, and the TF extracellular domain shares the cytokine receptor architecture of two fibronectin type III domains aligned in an elongated manner with an angled and rigid intramodule interface.31,32,33,34 However, the TF cytoplasmic domain lacks the typical cytokine receptor tyrosine kinase recognition motifs. Instead, a Ser/Thr-Pro phosphorylation site that is the target for unidentified Pro-directed kinases is highly conserved in all mammalian species.35 Phosphorylation of the canonical Ser258 in the human TF cytoplasmic domain requires prior protein kinase C-dependent phosphorylation of a more amino-terminal Ser-Xxx-Lys motif35,36,37 and is regulated by palmitoylation of a conserved membrane proximal Cys residue36,38 (FIGURE 44.1). The phosphorylated TF cytoplasmic domain has a more compact structure, and phosphorylation might thus prevent or promote the binding of intracellular adaptor proteins supporting TF trafficking.39
The TF extracellular domain mediates ligand interactions with each of the structural domains of VIIa. The docking of the protease domain leads to allosteric activation of catalytic activity of the zymogen-like protease VIIa,40 whereas interactions of the VIIa light chain with the carboxyl-terminal module of TF confer high-affinity binding and stable TF-VIIa complex formation necessary for efficient activation of macromolecular substrates X and IX. The details of these procoagulant interactions of TF have been elucidated at the molecular level.41,42,43,44 On cell surfaces, a substantial portion of TF is not involved in the initiation of coagulation, although these pools of TF bind VIIa and activate its catalytic activity. Procoagulant TF, as found on activated monocytes, has high affinity for VIIa (KD < 1 nM), but noncoagulant cell surface pools of TF are only saturated at higher concentrations of VIIa.45,46,47 Chemical cross-linking and the binding of substrate-dependent inhibitors48 or antibody reactivity49 indicate that the noncoagulant pools of TF have a distinct tertiary or quaternary structure. Heterogeneity of cellsurface TF is also evident from functional dose-response curves, demonstrating that higher concentrations (5 to 20 nM) of VIIa are required to saturate pools of TF involved in direct TF-VIIa signaling.49,50,51
Multiple factors contribute to the functional and structural heterogeneity of cell-surface-expressed TF. Negatively charged surfaces containing phosphatidylserine increase the measured
affinity of VIIa for TF in purified systems.52,53 The cellular lipid environments surrounding TF could alter TF structure54 or increase VIIa affinity through contacts of the VIIa Gla domain. TF is found in phospholipid-rich as well as glycosphingolipidrich raft microdomains and caveolae of the cell membrane. The dynamic trafficking of TF between subcellular locations is a critical determinant for TF procoagulant activity.55,56,57,58 TF cellsurface expression is regulated by basolateral sorting in endothelial and epithelial cells,59 and basolateral sorting and formation of cell-cell contacts influences cell-surface procoagulant activity and changes TF glycosylation.49,60
affinity of VIIa for TF in purified systems.52,53 The cellular lipid environments surrounding TF could alter TF structure54 or increase VIIa affinity through contacts of the VIIa Gla domain. TF is found in phospholipid-rich as well as glycosphingolipidrich raft microdomains and caveolae of the cell membrane. The dynamic trafficking of TF between subcellular locations is a critical determinant for TF procoagulant activity.55,56,57,58 TF cellsurface expression is regulated by basolateral sorting in endothelial and epithelial cells,59 and basolateral sorting and formation of cell-cell contacts influences cell-surface procoagulant activity and changes TF glycosylation.49,60
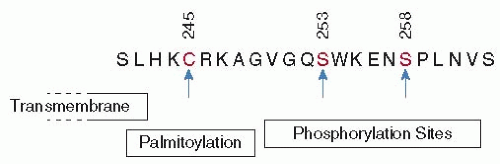 FIGURE 44.1 Features of the human TF cytoplasmic domain. |
Human TF has three extracellular N-linked glycosylation sites that to a variable degree determine the overall carbohydrate content of the molecule.61,62,63 Whereas secretion of recombinant soluble TF extracellular domain is markedly reduced by mutation of the most amino-terminal N-linked glycosylation site,61 overexpression of full-length TF in commonly used cell lines did not reveal reduced procoagulant activity following mutation of any of the TF glycosylation sites.63,64 However, biochemical comparison of highly purified TF from natural and recombinant sources demonstrated that recombinant TF has less complex, high mannose carbohydrates, while natural TF recovered from a particular thromboplastin reagent contains only hybrid, highly fucosylated, and sialylated sugars.65 Natural TF has higher specific procoagulant activity, indicating that these complex carbohydrates significantly modulate TF activity. Recent experiments in mice show that TF glycosylation either marks or is required for cell-surface expression of TF in epithelial cells in vivo. A rapid change in TF glycosylation in intestinal epithelial cells occurs following colonization of the small intestine with microbiota, and the resulting cell-surface expression of TF triggers coagulation, procoagulant PAR1 signaling, and adaptive angiogenesis.66
The carbohydrate composition of proteins is determined by intracellular trafficking and recycling routes. Ligand binding of VIIa to TF induces internalization and degradation, but a significant portion of procoagulant TF recycles back to the cell surface with its ligand VIIa associated.67 TF-VIIa endocytosis and the subsequent mobilization of additional pools of TF from the Golgi are regulated by the activation of PAR2.68,69 In addition, the TF-VIIa-Xa ternary coagulation initiation complex is inhibited on cells by TF pathway inhibitor (TFPI). TFPI is anchored to the cell surface by a glycosylphosphatidyl-inositol (GPI) lipid attachment, either by direct protein modification of the splice variant TFPIβ or potentially by binding to an unidentified GPI-anchored receptor in the case of TFPIα.56,70,71,72 TFPIβ is mainly responsible for regulating TF procoagulant function and represents the major GPI-anchored form in endothelial cells in culture.73,74 TFPI is coexpressed in most tissues with an androgen-induced, raft localized protein that regulates TFPI expression and inhibitory activity.75 GPI-anchored TFPI directs the TF ternary complex to caveolae and initiates a slow recycling pathway that eventually restores in part both TF coagulant activity and TFPI inhibitory control on the cell surface.56,76 Thus, binding of VIIa or formation of the ternary coagulation initiation complex are not static cell-surface events, but promote cellular trafficking and internalization of TF.
TF function in thrombosis and cell signaling is under the control of thiol-disulfide exchange pathways.49,77 In vitro and in vivo evidence supports roles for protein disulfide isomerase (PDI)-mediated thiol exchange in regulating TF procoagulant activity, microparticle release, and thrombosis.49,78,79,80,81 The TF extracellular domain has a surface exposed Cys186-Cys209 disulfide bond with a redox potential indicative of a functional, allosteric disulfide that can undergo reversible reduction and oxidation.77,82 Mutational breaking of this disulfide impairs procoagulant activity,83 but an intact disulfide is not required for direct TF-VIIa cell-signaling activity that is supported by a TF mutant with a single, unpaired Cys186 with a dose-response curve indistinguishable from wild TF.49 Precise mechanisms of breaking the allosteric disulfide on cells remain to be elucidated, and it is still debated whether thiol modifications of TF itself is the predominant regulator of cellular procoagulant function.84,85 Evidence that cell-surface PDI is associated with TF on cells expressing a TF-PAR2 signaling complex indicates that disulfide exchange is a cellular pathway that can switch certain cellular pools of TF to a selective signaling receptor.49
TF-SIGNALING COMPLEXES AND CORECEPTORS CONTRIBUTING TO PROTEOLYTIC PAR ACTIVATION
Groundbreaking studies by Hans Prydz and colleagues demonstrated that the upstream coagulation proteases VIIa and Xa elicit cell-signaling events that are dependent on their proteolytic activities, but independent of thrombin-mediated activation of PARs.86,87 VIIa in addition to TF-expressing cells activates several signaling cascades and transcriptional responses, as documented subsequently in various experimental systems.88,89,90,91,92,93 TF-VIIa signaling prevents apoptosis and anoikis (cell death following loss of adhesion).94,95 Several of these TF-VIIa signaling events, including the activation of mitogen-activated protein kinase (MAPK) pathways and the upregulation of kinase cascades promoting protein synthesis, were found to be independent of the TF cytoplasmic domain in overexpression systems.91,96
The intracellular signaling elicited by VIIa was reminiscent of thrombin signaling through the G protein-coupled receptor PAR1, but a candidate PAR receptor that is cleaved by TF-VIIa remained elusive.97 Reconstitution experiments clearly demonstrated that TF-VIIa signals through the thrombin-insensitive PAR2.98,99 Experiments with blocking antibodies or knockdown approaches confirmed the critical role of PAR2 as a signaling receptor for TF-VIIa in normal and transformed cells.51,100,101 Similarly, PAR2 emerged as the receptor that mediates signaling of Xa,102,103,104 but Xa also cleaves PAR1 directly and independent of thrombin generation.105 The structure-function analysis of PAR2 cleavage by TF-VIIa identified hotspots for binding of PAR2 in the VIIa catalytic cleft and enabled the design of PAR2 variants that are selectively activated by TF-VIIa, but not Xa.106 Thus, PAR2 is a common signaling receptor for upstream coagulation proteases of the TF pathway, but distinct coreceptors
further diversify the signaling responses induced by proteolytic activation via TF-VIIa or Xa (FIGURE 44.2).
further diversify the signaling responses induced by proteolytic activation via TF-VIIa or Xa (FIGURE 44.2).
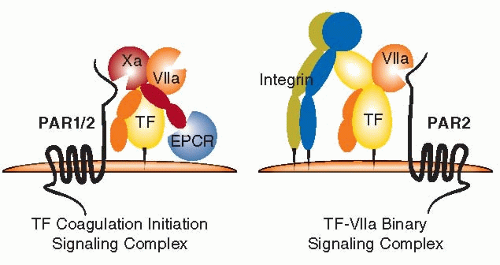 FIGURE 44.2 Schematic overview of the proteolytic TF-signaling complexes and associated coreceptors. |
The combined addition of VIIa and X to TF-expressing cells is a potent stimulus for PAR2 signaling that occurs concomitantly with coagulation activation.98,99 Although high concentrations of VIIa elicit cell signaling independently of Xa generation, it is the nascent product Xa in the high-affinity TF-VIIa coagulation initiation complex that primarily cleaves and activates PAR1 or PAR2.49,99 Cell-surface-anchored TFPI not only regulates the procoagulant function but also signaling of the ternary TF-VIIa-Xa complex via PARs.107 Thus, initiation of coagulation is mechanistically coupled with cell-signaling events and controlled by the physiologic inhibitor of the TF pathway. This principle similarly operates in the anticoagulant pathway in which the thrombomodulin-thrombin complex generates activated protein C (APC).108 Initiation of the anticoagulant pathway is supported by Gla domain-mediated binding of PC to the endothelial cell protein C receptor (EPCR),109 which subsequently supports APC-mediated cytoprotective PAR1 signaling.110,111
A broader role for EPCR is emerging, because EPCR binds not only APC but also VIIa with similar affinity,112,113 and EPCR has been implicated in Xa-mediated PAR1 activation in overexpression systems.114 The low affinity of X/Xa for EPCR relative to VIIa and APC115 suggested a preferred interaction of TF-VIIa with EPCR. However, mutations of the VIIa Gla domain that severely reduce EPCR binding were without effect on PAR cleavage by either the binary or the TF-VIIa-Xa ternary complex.116 In contrast, signaling of the ternary TF-VIIa-Xa complex was strictly dependent on EPCR, in both human and mouse cells,116,117 indicating that EPCR is involved in functional interactions with Xa, rather than VIIa. These initial data indicate that EPCR is a cosignaling receptor for both procoagulant and anticoagulant protease complexes, a concept that requires further exploration in vivo.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


