Clinical Manifestations and Therapy of Inherited and Acquired Hemophilia
Edward G.D. Tuddenham
Peter W. Collins
T. T. Yee
HISTORICAL INTRODUCTION
Knowledge of a congenital bleeding disorder affecting male infants can be inferred from rulings in the Babylonian Talmud1 compiled about 500 CE. A dispensation was given to forgo circumcision in brothers and cousins of affected boys who had died after the ritual was carried out (presumably from bleeding), showing that the Rabbis recognized inheritance of a bleeding tendency through carrier females. Abu al-Qasim Khalaf ibn al-Abbas Al-Zahrawi, known as Albucasis (936-1013), the great Moorish surgical writer of 10th-century Spain, wrote of a certain village where boys and men suffered excessive bleeding after minor injury. He also gave the first practical advice for treating such bleeding by means of cautery, the success of which he observed. We can now interpret this report as referring to moderately severe hemophilia with a local founder effect.
The curious word for an abnormal inherited bleeding tendency which we use today—hemophilia (literally blood lover)—was introduced by Hopff in his doctoral thesis delivered at Wurzburgin 1828.
The medical literature of the 18th and 19th centuries on hemophilia was gathered in a monumental review published in 1911 by the Francis Galton Laboratory for National Eugenics of the University of London (now abbreviated to The Galton Laboratory). The review2 described clearly all the clinical features that are seen today in untreated cases of hemophilia. However, they failed to recognize the key feature of X-linked inheritance—daughters of affected males are obligate carriers of the mutant gene. The correct theoretical explanation of chromosomal sex determination and of sex-linked inheritance in mammals had only just been proposed and had evidently not come to their notice. Notably absent from the nearly 1,000 pedigrees of hemophilic families illustrated in the review was that of the most famous hemophilic family in history, namely the descendants of Queen Victoria, who was an obligate carrier on pedigree evidence (FIGURE 51.1). In addition to her British descendants, she transmitted hemophilia to three other European Royal families through dynastic marriage alliances. No antecedents of Queen Victoria had a bleeding tendency, so it is likely that she inherited a new mutation in one or other of the parental gametes, as is the case for a third of carriers of hemophilia. Three of her daughters were carriers of the hemophilic gene and her youngest son Leopold had clinically severe hemophilia. The mutant gene then made its way into the German, the Spanish, and most famously the Russian Royal families. Leopold’s daughter, the Princess Alice, was an obligate carrier and had two affected sons who died in childhood. The German affected boys also died young, but the Spanish princes with hemophilia survived into their 20s before succumbing to trauma-induced bleeding. Alexey Romanov, the Tsarevich (heir to the throne of Russia), was affected with severe hemophilia. Alexey was murdered on Lenin’s orders with his family at Yekaterinburg during the civil war following the Bolshevik revolution. Hence by 1938, there were no surviving male descendants of Queen Victoria with hemophilia. In 1952, it emerged that there are two types of hemophilia—A and B. The question then arose, whether the “Royal disease” was type A or type B. It became a long-standing historical puzzle which was finally solved almost 60 years later, by a tour de force of forensic genetics, in favor of hemophilia B,3 by genomic analysis of the Imperial family’s bones, recovered from unmarked graves near Yekaterinburg (FIGURE 51.2).
GENETICS AND BIOCHEMISTRY OF HEMOPHILIA
The X-linked inherited bleeding disorders, hemophilia A and B, are due respectively to mutations at the factor VIII (FVIII) locus (f8) or the FIX locus (f9), both situated on the long arm of chromosome ×X at Xq2.8 and Xq2.6, respectively. Thef8-linked condition, caused by deficient synthesis of coagulation FVIII, is known as hemophilia A, whilst the f9-linked condition, due to deficient synthesis of coagulation FIX, is called hemophilia B or Christmas disease (after one of the first described sufferers). The clinical features of the two conditions are indistinguishable, and they were not separated until complementation studies in 1952 uncovered the fact that blood from a majority of patients with X-linked hemophilia (type A) could correct the coagulation defect in blood from a minority (type B) and vice versa. FIX itself was discovered as a result of these studies, being called at first Christmas factor4 to distinguish it from antihemophilic factor, the name already assigned to the putative entity lacking in the blood of type A patients. Biochemically, FVIII and FIX function together as a binary enzyme (FIXa) cofactor (FVIIIa) complex whose substrate is FX.
The incidence ratio of hemophilia A to B is about 5 to 1, and both conditions are rare: hemophilia A affects 1 in 5,000 males at birth whereas hemophilia B affects only 1 in 25,000 males at birth. Contrary to the commonly held belief that carriers are unaffected, about a third of females who are heterozygous for mutations at either locus manifest mild hemophilia due to skewed ×X inactivation.
Acquired hemophilia A is a rare autoimmune disorder caused by failure of self-tolerance toward FVIII occurring in later life, with production of antibodies neutralizing the activity of the patient’s own FVIII. Hemophilia A and B are treated together in the following discussion of the pathology, clinical features, and treatment of the two disorders, only noting differences where relevant.
 FIGURE 51.1 Queen Victoria’s pedigree. The Royal disease is an ×X-linked recessive bleeding disorder that, until resolved by modern genetic analysis, could have been either hemophilia A or B as every affected male had died before blood tests could have decided the issue in 1950. Obligate carriers on pedigree are represented by a circle with a red dot in its centre. Affected males are shown as red squares. Of the four royal families afflicted by the disease (blue circles), the most tragic was the Russian. Ale×xei met his end by murder as did his sisters and parents. One of his sisters was a carrier according to the forensic analysis, probably Anastasia (FIGURE 51.2). (Reproduced from Lannoy N, Hermans C. The “royal disease”—haemophilia A or B? A haematological mystery is finally solved. Haemophilia 2010;16(6):843-847, with permission.) |
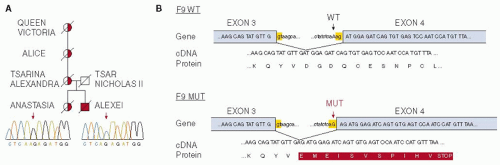 FIGURE 51.2 The Royal disease was caused by a point mutation in F9. A: Partial pedigree of the Royal family, showing transmission of the mutation from Queen Victoria to Empress Alexandra and from Alexandra to Prince Alexei, her haemophilic son. Alexei was hemizygous for the mutation, whereas Alexandra and one of her daughters, putative Grand Duchess Anastasia, were heterozygous carriers. B: The A-to-G mutation occurs just upstream of exon 4 in the F9 gene and is predicted to create a new splice acceptor site that could lead to production of a truncated factor IX protein. WT indicates wild type. (Reproduced from Rogaev EI, Grigorenko AP, Faskhutdinova G, et al. Genotype analysis identifies the cause of the “royal disease”. Science 2009:326(5954):817, with permission.) |
PATHOPHYSIOLOGY AND PRINCIPLES OF TREATMENT
All clinical features of hemophilia A and B are due to deficiency of FVIIIor FIX respectively, with consequent failure to generate adequate thrombin in response to injury and hence failure to form a stable hemostatic plug at sites of vascular damage. Correction of the defect by intravenous infusion of the relevant missing factor completely normalizes thrombin generation, since all other factors required for hemostasis are present in normal amounts. Several circumstances conspire to frustrate our efforts to normalize clotting in a hemophilic person’s blood. FVIII and FIX are scarce proteins which are difficult to purify and difficult to make by genetic engineering. Hence, the cost of clinical factor concentrates is extremely high. Given a supply of factor concentrate, it still has to be introduced into the circulation by intravenous infusion. Vascular access is therefore critical for hemophilia care lifelong. Most problematic of all is the short half-life in the circulation of infused factor— 8 to 12 hours for FVIII and 15 to 20 hours for FIX. In fact, four out of five hemophiliacs worldwide have little or no access to replacement therapy at all. Treatment regimens discussed below are a compromise designed to maximize the benefit of scarce resources and minimize the dire effects of bleeding, which if left unstaunched leads to progressive damage wherever bleeding is prone to occur.
If left without effective treatment the natural history of hemophilia is progressive loss of mobility from joint and muscle bleeding and early death due to exsanguination or uncontrolled internal bleeding, most often intracranial. Prior to the advent of factor concentrates in the 1970s, the most severely affected hemophiliacs died before the age of 20. In contrast to that dire prognosis, modern comprehensive hemophilia care, supported by the resources of an economically advanced state, can normalize life span and limit or eliminate tissue damage.5 Adequate supplies of virus safe FVIII and FIX first became widely available in the 1990s; hence most hemophiliacs born before 1985, even in economically advanced countries, are living with the long-term effects of undertreatment in childhood and infection by hepatitis C and HIV, which were present in factor concentrates up to 1986.
GENETICS AND DIAGNOSIS OF HEMOPHILIA AANDB
Since there is no heterozygote advantage to carrying defects at the f8 or f9 loci and since the hemizygous male has severely limited reproductive fitness (absent treatment), the frequency of “hemophilic” gene defects in the population must be maintained by new mutations. Consequently and as predicted by Haldane (op. cit.), the mutations are highly diverse.
Usually genetic analysis is carried out following the measurement of blood factor level establishing the diagnosis in the affected male, but putative carriers of hemophilia A or B often have no living affected relative, due to early death. Alternatively they may be carriers of a recent mutation. To help such women with establishment of carrier status and genetic counseling, it is then necessary to sequence their f8 and/or f9 genes. Detection of a heterozygous mutation already noted in the databases associated with hemophilia, or (if not previously reported) plausibly causative of deficient/functionally defective FVIII or FIX, then confirms carrier status. The nature of the mutation can also predict the severity of the disorder. Severe gene lesions such as stop codons, large deletions, or the prevalent inversion in hemophilia A not only cause severe disease with no circulating factor but are also associated with high risk of neutralizing antibody (inhibitor) development after substitution therapy. Missense mutations on the other hand are usually associated with moderate (residual factor level 1% to <5%) or mild (residual factor > 5%) bleeding tendency.
In laboratory diagnosis of hemophilia A and B, the standard screening tests show a prolongation of activate partial thromboplastin time (APTT) with normal prothrombin time (PT). If this is the case, then specific factor assays to quantify FVIII and FIX are performed. Traditionally, residual FVIII or FIX <1% is defined as severe. However, the lower detection limit for both assays is about 1% and a residual level below 1% but above absolute zero can make some difference to bleeding frequency, that is to say—the clinical phenotype. This is notable when one compares the bleeding frequency of subjects with a null mutation against those with a missense mutation but factor level <1% who may even have no spontaneous bleeding.6 Although uncommon, patients with multiple defects in their hemostatic system are also found.
An assay capable of detecting antibodies to FVIII or FIX is also a mandatory part of the laboratory workup. In hereditary hemophilia A and B antibodies only develop after treatment and may do so promptly thereafter. Inhibitors may develop after many years of treatment, as recent data from the UK national database show.7 Thus, regular inhibitor testing is part of routine follow-up care in hemophilia.
There are two different ways of measuring FVIII activity, the one-stage clotting assay and the two-stage clotting assay. Ideally, both assays should be performed on all patients because many patients with mild hemophilia A have a much lower two-stage assay level than one-stage assay level. The clinical bleeding tendency is better predicted by the two-stage assay result. There are even a few patients who have normal one-stage assay result but clearly abnormal two-stage assay.8
THE CLINICAL PHENOTYPE IN HEMOPHILIA AANDB
A guide to bleeding frequency can be derived from the residual factor level in the patient’s blood9 (FIGURES 51.3A,B). Subjects whose residual level is <1% can attract clinical attention at birth, with bleeding complications of delivery or within a few months with spontaneous bruising, bleeding after circumcision, from primary dentition or hemarthrosis. Throughout life, such patients bleed apparently spontaneously, but also after minor trauma about once or twice a week in the most severe cases.
Patients whose residual level is 1% to <5% are said to have moderate hemophilia. Typically, they present at age 2 to 5 years with bleeding after significant trauma that has sufficiently intrigued a medical attendant to request a blood test including screening for coagulation disorders. If the residual level is over 5%, then the patient is said to have mild hemophilia. Bleeding only occurs in such cases after significant trauma or surgery. Patients with mild hemophilia are often first diagnosed in middle or late life and then only after severe trauma or major surgery brings the bleeding tendency to light. The diagnosis can
go undetected for many years; as in one extreme example noted by one of us (ET), a 90-year-old gentleman was found to have 5% FIX that came to light because he bled around the site of catheter insertion for coronary angiography.
go undetected for many years; as in one extreme example noted by one of us (ET), a 90-year-old gentleman was found to have 5% FIX that came to light because he bled around the site of catheter insertion for coronary angiography.
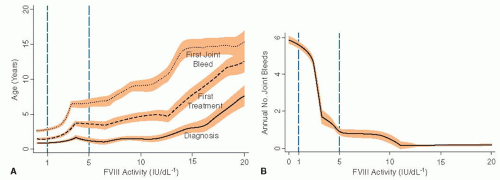 FIGURE 51.3 A: Age at diagnosis, onset of treatment, and joint bleeding according to factor VIII activity in a cohort of haemophilia A patients followed at one centre in the Netherlands (Ellis van Kreveld haemophilia centre). Lines are medians, shaded areas are interquartile range. (Reproduced from Den Uijl IE, Mauser Bunschoten EP, Roosendaal G, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia 2011;17(6):849-853, with permission.) B: Annual number of joint bleeds according to factor VIII activity in the same cohort as in FIGURE 51.3A. Of note, there were no joint bleeds in patients with a baseline level above 12 lU/dL (Reproduced from Den Uijl IE, Mauser Bunschoten EP, Roosendaal G, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia 2011;17(6):849-853, with permission.) |
The anatomical structures most frequently affected by hemophilic bleeding are the major load-bearing joints in the order of ankles>knees>elbow>shoulders. The hips are often spared. A target joint is one where repeated bleeding is occurring, without fully recovering between bleeds. Such joints go on to develop progressive painful, deforming, and motion-restricting arthropathy until they become fixed by reactive fibrosis. Blood within a joint capsule is highly irritant and provokes proliferative synovitis with erosion of cartilage, formation of bone cysts and deformity, and progressive fibrosis.10 The progress of such damage to joints is monitored for purposes of assessing response to treatment by standardized scales based on range of movement and function, radiology, or MRI.11,12,13,14
Muscles develop bleeding after direct trauma or exercise or spontaneously. If the blood is confined within a muscle capsule, pressure can build up until compartment syndrome develops. This is a surgical emergency since it can lead to ischemic necrosis and contracture with loss of all function. An incision in the fascia surrounding the affected muscle combined with adequate replacement therapy is muscle saving in such circumstances, though not without risk of continued bleeding.
Bleeding into deep muscles such as the iliofemoral muscles can occur in all severity levels of hemophilia. Hip flexion, abdominal pain, and femoral nerve entrapment symptoms should lead to suspicion of this site of bleeding.
Renal tract bleeding presenting with hematuria is common and often not related to any detectable renal tract disease.
Intracerebral bleeding has been the commonest cause of death in hemophilia, even to the present.
Bruising is commonly observed in hemophilic children on the legs, trunk, and arms but is not usually a cause of concern and does not need treatment if superficial and painless.
THE SEVEN AGES OF HEMOPHILIC MAN
Conception
The ethical choice not to have a hemophilic child is one that many carriers have made, in view of their first-hand observation of the suffering of their father or (N.B. not “and”) brothers. Recently, preimplantation genetic diagnosis has made it possible to select embryos, obtained by in vitro fertilisation (IVF), for transfer according to their genetic status, and this technique has been successful for hemophilia.15
In Utero
Prenatal diagnosis, undertaken either to terminate an affected conceptus or to guide subsequent management of labor and delivery, is widely practiced by means of chorion villus biopsy. This allows direct access to tissue and therefore DNA of fetal origin, at 10 to 12 weeks, which can be tested for linkage to or directly for the specific mutation present in the family of the carrier.
Noninvasive testing for fetal DNA present in maternal blood has been developed using digital quantitative polymerase chain reaction (PCR).16 From as early as the 11th week of gestation, it is now possible to determine by analysis of maternal blood not only the sex of the conceptus but also whether it is or is not affected. When the fetus is affected, the quantity of DNA bearing the mutation in the maternal plasma is increased above the level due to the maternal heterozygosity in a ratio that is high enough to enable perfect discrimination from the ratio when the male
fetus is unaffected. Either the specific mutation or a linked informative single-nucleotide polymorphism may be used.
fetus is unaffected. Either the specific mutation or a linked informative single-nucleotide polymorphism may be used.
In utero bleeding is extremely rare in a hemophiliac fetus, suggesting that it is highly protected against trauma whilst in the womb.
Delivery of Affected Males
Passage through the birth canal of a term infant can only be achieved with considerable compression of the skull and deformation of the brain. At least 5% of hemophilic infants develop clear neurological signs of progressive bleeding requiring treatment. If the diagnosis of hemophilia has been made already, then the baby can be closely monitored and treated at the first sign of such bleeding. Tragedies occur when the diagnosis has not been expected, which is true of about one-third of such deliveries due to new mutations (see section on Genetics above). Current guidelines advise against prolonged labor, instrumental delivery, and use of scalp monitoring.17 There should be a low threshold to proceed to cesarian section if labor is not progressing and difficulties in the birth canal are likely. A sample of cord blood should be taken to confirm or exclude hemophilia. FVIII may be boosted by the stress of birth such that an elevated level may be present in mild but not in severe hemophilia. FIX levels are reduced in normal term and more so in preterm cord blood. However, absent or very low levels are clearly indicative of hemophilia B.
The Infant
In neonates with severe hemophilia, the decision to infuse prophylactic concentrate at birth is controversial. Some authorities recommend replacement therapy without evidence of active bleeding whilst others wait for evidence of bleeding. The recommended practice in Europe and North America is to delay the start of regular prophylaxis until the infant starts to crawl and has his first joint or muscle bleed. This will vary from 12 to 18 months. To secure venous access, an indwelling port may need to be inserted and the parents trained in its use so that a regular program of prophylaxis can be maintained. This standard of treatment is now producing for the first time children and young men with severe hemophilia who have never had a serious bleed and who have normal joints. However, there is now also an increased incidence of inhibitory antibody development in children with severe hemophilia A, occurring within the first 50 infusions.
The School Years
Since the practice of early and continuous prophylaxis was instituted, hemophilic children have been able to attend mainstream schools and take part in all activities with the exception of violent contact sports. With maturity, the hemophilic child is encouraged to take responsibility for his condition and to self-administer concentrate. Most children with hemophilia can achieve this by the age of 10.
Adolescence and Transition to the Adult Center
There comes a time when the boy is reaching adolescence and should have his care transferred to an adult centre. When this time is reached varies, as does the timing of puberty. Special sensitivity to the patient’s and parents’ needs will ease this transfer, which should be managed formally according to a scheme that ensures none of the patient’s needs are overlooked. Risk taking may manifest during adolescence, but if good habits of treatment and prophylaxis have been ingrained in childhood, then most young men with hemophilia can cope with the added stress of the teen years without losing control of their hemophilia.
Early Adulthood and Career
Hemophilia is no longer a bar to any level of educational attainment. Nor is it a bar to entry to any trade or profession, with the exception of front-line military and police work. No doubt, those bars too will be lifted one day, either by very long-acting preparations or by gene therapy (see section on Gene Therapy below).
Reproductive fitness of hemophilic males is now similar to that of other males. However, the question of inheritance does arise and is best dealt with in the context of a genetic counseling clinic with trained counselors, geneticist, and hematologist, roles that may be combined in one or two people. The obligate carrier daughter will herself in due course need genetic counseling. Numbers of carrier females and of affected male infants born to them are on the rise, thanks to improved therapy.
Middle Years
Men with severe hemophilia who have survived to the age of 35 and over have all been infected with hepatitis C, and most have also been infected with HIV in childhood. They may also have badly damaged joints with progressive arthropathy due to inadequate on-demand treatment of bleeding in their early years. Their medical needs contrast dramatically with those of the younger well-treated generations now reaching maturity. In those who have failed to clear hepatitis C, there is the potential for progression to cirrhosis with increasing risk of hepatocellular carcinoma and of terminal liver disease. These patients benefit from liver transplantation as the last resort, which also cures hemophilia A and B. Multiply disabled hemophiliacs (older men in developed countries and younger subjects from developing nations) depend for their mobility on the help of skilled orthopedic surgeons to replace joints and specialized physiotherapists to maintain function.
Later Life
For the first time in history, significant numbers of men with severe hemophilia are reaching their 6th, 7th and even 8th decades. A particular puzzle arises in relation to arterial degenerative disease and hemophilia. Although the rate of coronary thrombosis is reduced in hemophilia, due to the permanent state of anticoagulation that is characteristic of the condition, arterial disease itself continues unabated and often leads to the need for arterial stenting or bypass with subsequent requirement for antiplatelet agents. The compromise that may be recommended is to use lower doses of antiplatelet agent whilst maintaining prophylaxis of the hemophilia at a slightly higher level than usual for the patient.18,19
Old Age and Death
Whilst hemophilic life span, in those who are not subject to the dire effect of chronic viral infection, or of resistant inhibitors, is now approaching the norm for the general population, there still remains an excess of deaths due to intracerebral hemorrhage.5 Prophylactic regimens do not deliver a normal level of FVIII or FIX all or even most of the time. Therefore, all patients spend most of their lives with subnormal thrombin generation.
Perhaps, the aging arteries in the brain, which become more fragile with age, finally start to leak, and the absence of adequate hemostasis at that time and place is fatal.
Perhaps, the aging arteries in the brain, which become more fragile with age, finally start to leak, and the absence of adequate hemostasis at that time and place is fatal.
One British hemophiliac has died who was found to have variant Creutzfeldt-Jakob disease (vCJD) prion in his spleen postmortem,20 as a result of receiving concentrate made from blood products contaminated with the agent. All hemophiliacs who received concentrate made from UK plasma in the relevant period are deemed at risk to develop or transmit vCJD.
TREATMENT OF UNCOMPLICATED HEMOPHILIAAANDB
Mandatory normal hemostasis must be secured for treatment of head injury/intracranial hemorrhage, major trauma, impending compartment syndrome, exsanguination, and surgery. Normal hemostasis, that is, FVIII or FIX level maintained at 100% ± 5%, is most securely and economically achieved by continuous infusion. High-purity plasma-derived and recombinant factors may be used in continuous infusion; for this, purpose a suitable dose is made up and delivered by syringe pump at a rate of about 1 U/kg/h after a bolus dose has been delivered to raise the level rapidly to 100%. Interval assay levels, throughout the period where normal hemostasis is required, should be used to adjust the infusion rate.
Undertreatment is the commonest mistake made in hemophilia care. An example of the consequences of undertreatment may be seen in the recent analysis of outcomes of knee joint replacement.21 Centers that covered surgery with normalization of factor level for 2 weeks followed by gradual reduction had a zero complication rate and centers using less factor had poor outcomes in up to 20% of patients. This is in keeping with laboratory studies in hemophilic mice.22
Formulae for Calculating Bolus Doses to Achieve 100%
FVIII dose in units = patients weight in kg × percent rise required to reach 100% × 0.5
FIX dose in units = patients weight in kg × percent rise required × 1.0
The same formulae are used to calculate any other desired target level as suggested in treatment plans below.
On-Demand Treatment of Joint and Other Bleeds
Treating a bleed at the earliest opportunity when it occurs is referred to as on-demand treatment. Provided the patient has his supply of treatment at home and can self-administer the drug intravenously, bleeding is minimized and the rate of joint damage progression is slowed. Due to lack of supply, on-demand treatment made available at a medical centre is the norm for severely affected patients in most of the world, although this is clearly a suboptimal approach to treating hemophilia.23 Moderate and mildly affected patients are managed mainly with on-demand treatment. Self-therapy is always preferable as it enables the patient to take charge of his own treatment and eliminates delays. A general rule for hemophilia is “when in doubt treat with factor.”
Dosage recommended for on-demand treatment is largely empirical with little in the way of trial data to support particular regimens. A common policy is to give a fairly small dose for an uncomplicated joint bleed if treated early and to follow with a second dose in 6 hours if the problem is not resolving rapidly. In hemophilia A, that would be a dose of 15 to 30 U/kg body weight. In hemophilia B with the somewhat lower recovery, 20 to 40 U/kg is suggested. For a more severe bleed induced by trauma or not responding to a lower dose of therapy, the recommended range is increased to 20 to 40 U/kg (hemophilia A) or 30 to 60 U/kg (hemophilia B).
Prophylactic Treatment for Hemophilia
The aim of prophylaxis is to convert the individual from a severe to a moderate bleeding phenotype and so decreasing or preventing musculoskeletal damage.24 Prophylactic treatment has been shown to prevent joint damage and musculoskeletal disability in long-term retrospective cohort studies.25,26,27 A prospective randomized clinical trial of prophylaxis versus enhanced on-demand therapy in young children showed reduced radiological joint damage in the prophylaxis arm.23 On the basis of these studies, prophylaxis is now the recommended treatment for children with severe hemophilia and is being increasingly used in adults.28 In addition to its proven role in reducing arthropathy, it is likely that prophylaxis reduces the risk of other severe bleeds, such as intracranial hemorrhage.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


