TABLE 6.1 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
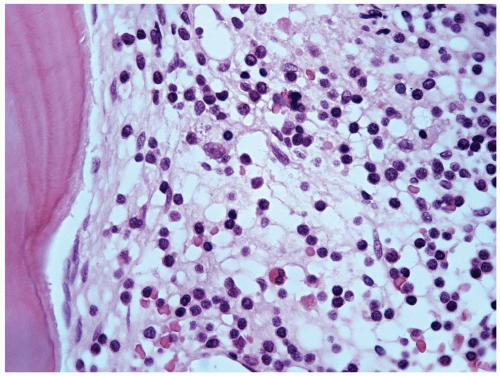 FIGURE 6.1 Bone marrow biopsy from a patient with acute myeloid leukemia after chemotherapy. Note a homogeneous population of small lymphocytes representing hematogones, as proved by flow cytometry. Hematoxylin and eosin, 60× magnification. |
the late-appearing T-cell markers (CD3 and CD5) may be decreased. In contrast, the late-appearing marker (CD3) is more frequently seen in peripheral T-cell lymphomas, and the early-appearing marker (CD7) may be absent. The gradual decrease in CD7+ cells in contrast to the persistence of CD3+ cells in cases of mycosis fungoides is one of the most dramatic examples. However, one must distinguish cytoplasmic from surface CD3, because the former is a marker that appears in immature T cells.
TABLE 6.2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The activation antigens are usually present on actively proliferating tumor cells, thus conferring a poor prognosis. Some activation antigens may be associated with a particular tumor, such as CD25 in hairy cell leukemia and CD30 in anaplastic large cell lymphoma.
saved in the refrigerator for additional monoclonal antibody testing, in the event that the preliminary diagnosis is incorrect or further subclassification is desired. However, Nguyen et al. (29) found that morphologic misinterpretation often occurred, leading to incorrect panel selection. These authors suggested setting up two large standard panels, designated blood/bone marrow/spleen panel and tissue/fluid panel, respectively.
TABLE 6.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 6.4 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
(Fig. 6.1.3) (5, 6 and 7). However, these cells are usually more immature than those in the blood, showing more promyelocytes and myelocytes. Blasts are usually <5% in the bone marrow. Myelodysplastic changes are not seen in the chronic phase. Megakaryocytes are increased and are characteristically microcytic and hypolobated (Figs. 6.1.4 and 6.1.5). In some cases, megakaryocytic hyperplasia is so prominent that some authors designated it Ph’-positive essential thrombocythemia (7). The degree of myelofibrosis is usually proportional to megakaryocytosis, and reticulin fiber
is increased in up to 40% of CML patients (5). Erythrocyte precursors are generally decreased, resulting in an M/E ratio as high as 10:1. Sea-blue histiocytes and pseudo-Gaucher cells (Fig. 6.1.6) are frequently present because of an increase of cell turnover. The bone marrow biopsy shows marked hypercellularity with granulocytosis and megakaryocytosis (Fig. 6.1.7). The immature myelocytes normally reside along the bony trabeculae and form a 2- to 3-cell layer. In CML cases, the paratrabecular cuff of immature myelocytes may become widened to up to a 5- to 10-cell layer (5).
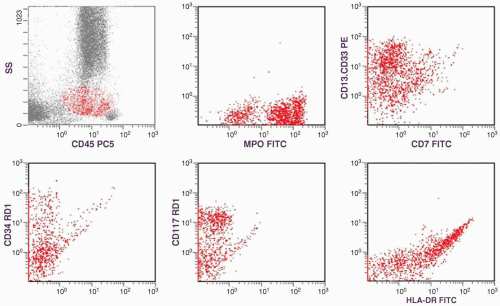 FIGURE 6.1.1 Flow cytometric analysis shows positive myeloperoxidase and CD13-CD33; partially positive CD34, CD117, and human leukocyte antigen (HLA)-DR reactions; but negative CD7 reaction. FITC, fluorescence in situ hybridization; PE, phycoerythrin; MPO, myeloperoxidose. |
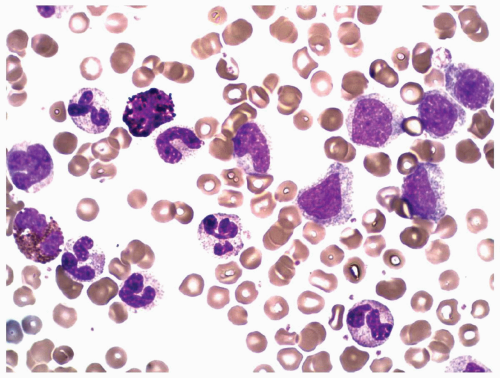 FIGURE 6.1.2 Peripheral blood smear from a patient with chronic phase of chronic myelogenous leukemia (CML) shows predominantly myelocytes and segmented neutrophils. A basophile and an eosinophil are also seen. Wright-Giemsa, 100× magnification. |
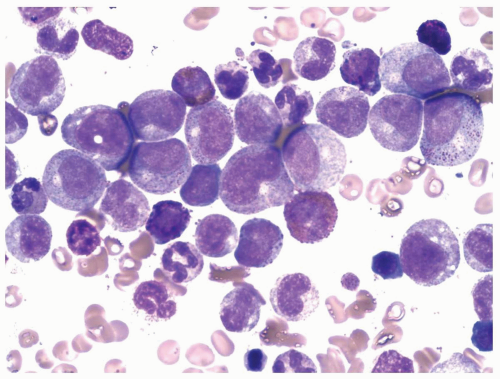 FIGURE 6.1.3 Bone marrow aspirate from a patient with chronic phase of chronic myelogenous leukemia (CML) shows predominantly promyelocytes and myelocytes with a few eosinophils and basophils. Wright-Giemsa, 100× magnification. |
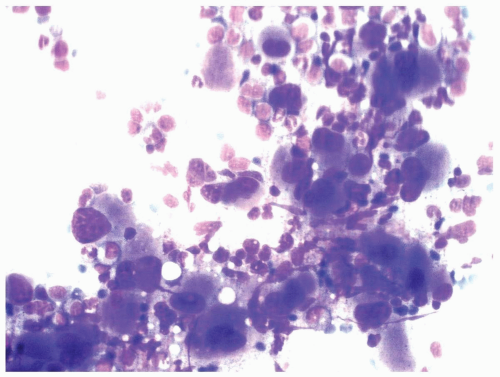 FIGURE 6.1.4 Bone marrow aspirate from a patient with chronic myelogenous leukemia (CML) shows a large cluster of megakaryocytes. Most megakaryocytes are mononucleated or hypolobated microcytic forms. Wright-Giemsa, 100× magnification. |
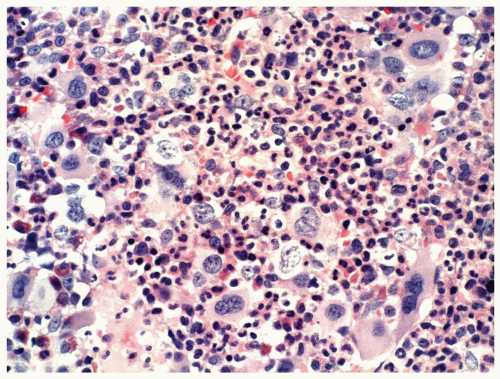 FIGURE 6.1.5 Bone marrow biopsy from a patient with chronic myelogenous leukemia (CML) reveals megakaryocytic proliferation. Hematoxylin and eosin, 40× magnification. |
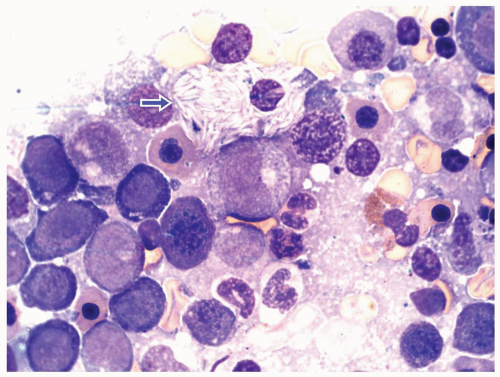 FIGURE 6.1.6 Bone marrow aspirate from a patient with chronic myelogenous leukemia (CML) reveals a peudo-Gaucher cell (arrow). 100× magnification. |
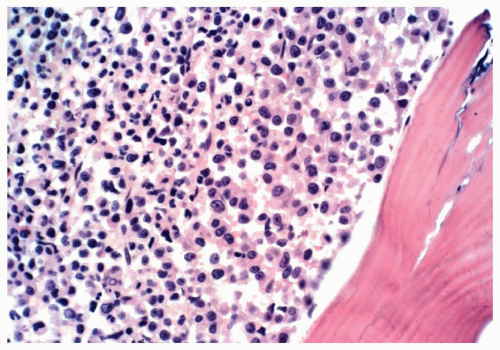 FIGURE 6.1.7 Bone marrow biopsy from a patient with chronic phase of chronic myelogenous leukemia (CML) shows widening of the paratrabecular cuff of immature myeloid cells. Hematoxylin and eosin, 40× magnification. |
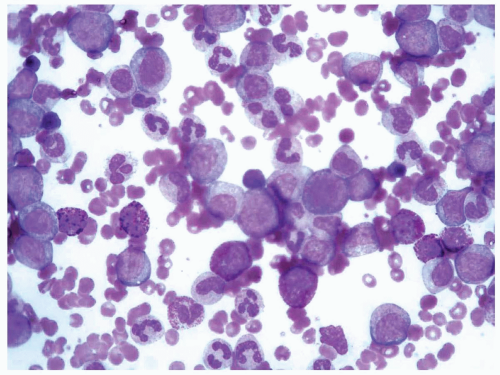 FIGURE 6.1.8 Peripheral blood from a patient with accelerated phase chronic myelogenous leukemia (CML) reveals an increased number of blasts with the presence of eosinophils and basophils. Wright-Giemsa, 100× magnification. |
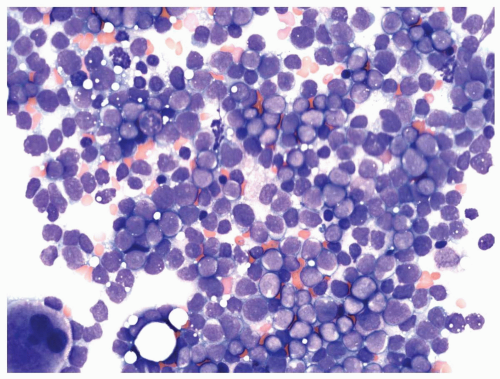 FIGURE 6.1.9 Bone marrow aspirate from a patient with blast phase chronic myelogenous leukemia (CML) shows 90% of blasts. Wright-Giemsa, 40× magnification. |
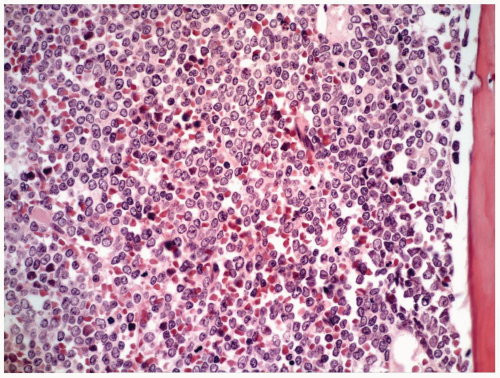 FIGURE 6.1.10 Bone marrow biopsy from a patient with blast phase chronic myelogenous leukemia (CML) shows total replacement of normal hematopoietic cells by the blasts. Hematoxylin and eosin, 40× magnification. |
different. The megakaryoblasts can be identified by CD41, CD42, and CD61, and the erythroblasts can be identified by glycophorin A and hemoglobin A staining.
blood and bone marrow, but the bone marrow should reveal prominent myeloid dysplasia. Chronic myelomonocytic leukemia may also show similar features, but there should be high monocyte counts in both peripheral blood and bone marrow with marked myelodysplastic changes.
TABLE 6.1.1 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
labor-intensive and difficult to standardize. Therefore, they have been gradually replaced by the quantitative real-time PCR techniques, using either the TaqMan or LightCycle system (28,29).
TABLE 6.1.2 | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
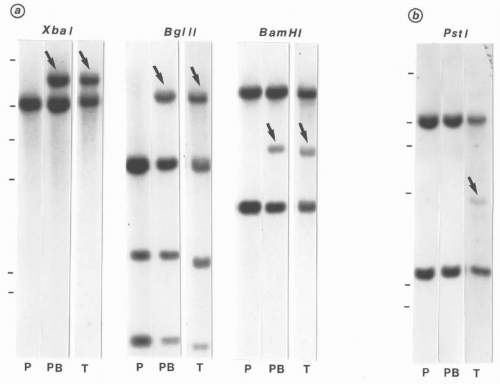 FIGURE 6.1.11 A: Southern blot hybridization analysis of DNA from peripheral blood (PB) and tumor tissue (T) with transprobe-1 (for breakpoint cluster region [BCR] or Philadelphia chromosome) showing identical rearrangement bands (arrows) after digestion with XbaI, BglII, and BamHI. P, placental control. B: Same tissues reacted with T-cell receptor β chain probe showing a faint rearranged band (arrow) in tumor tissue (T) after PstI digestion. (From Sun T, Susin M, Koduru P, et al. Extramedullary blast crisis in chronic myelogenous leukemia. Cancer. 1991;68:605-610, with permission.) |
phase. However, the recent use of BCR/ABL tyrosine kinase inhibitors has revolutionized the treatment of CML (15,30). The most commonly used drug is imatinib mesylate with the trade names of Gleevec in the United States and Glivec in Europe. It competitively binds to the adenosine triphosphate (ATP)-binding site of the BCR-ABL, and inhibits protein tyrosine phosphorylation (31). Imatinib mesylate induces complete cytogenetic responses in up to 90% of patients and major molecular responses in most of them (30).
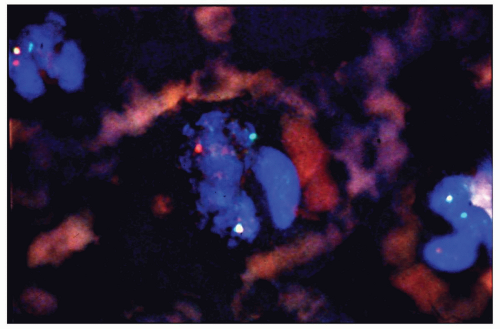 FIGURE 6.1.12 Fluorescence in situ hybridization of peripheral blood from a chronic myelogenous leukemia (CML) patient reacting with breakpoint cluster region (BCR) and Ableson (ABL) probes, showing a green signal (BCR), a red signal (ABL), and a yellow signal, which represents the overlapping of a green and red signal, in all three segmented neutrophils, indicative of BCR-ABL gene fusion product. |
manifested as the changes of the configuration and lobulation of the nuclei, the size of the nuclei and of the entire cell, and cytoplasmic granularity.
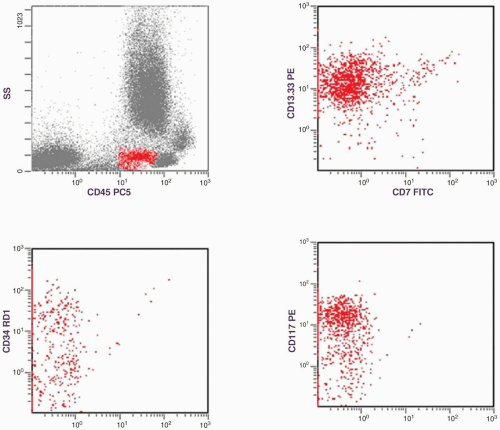 FIGURE 6.2.1 Flow cytometric histograms show the gating of an immature myeloid population with positive reactions to CD13.CD33, CD34, and CD117. This cluster represents the myeloblasts in a case of refractory anemia with excess blasts. |
TABLE 6.2.1 | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
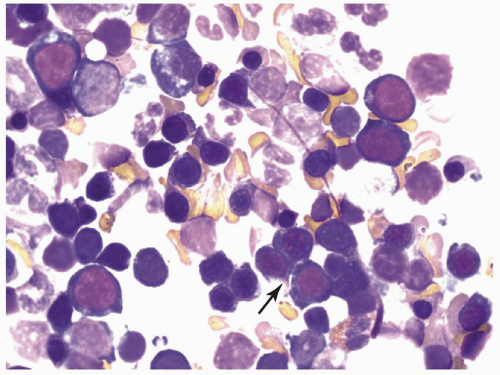 FIGURE 6.2.2 Bone marrow aspirate from a case of refractory anemia shows erythroid hyperplasia with the presence of a cluster of megaloblastoid normoblasts (arrow). Wright-Giemsa stain, 100× magnification. |
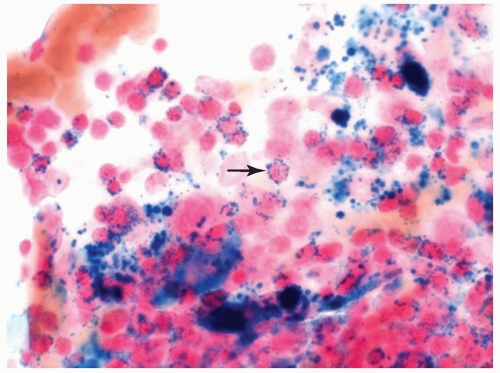 FIGURE 6.2.3 Bone marrow aspirate from a case of refractory anemia with ringed sideroblasts shows many ringed sideroblasts (arrow) in the Prussian blue-stained smear. 100× magnification. |
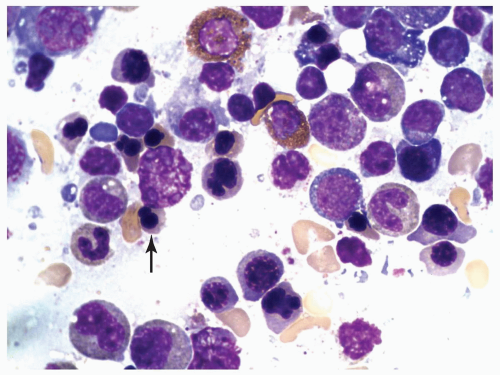 FIGURE 6.2.4 Bone marrow aspirate from a case of refractory anemia with excess blasts shows many dysplastic normoblasts with nuclear budding or bizarre nuclear shapes (arrow). Wright-Giemsa stain, 100× magnification. |
cases. The presence of pseudo-Chediak-Higashi granules has been reported, but this finding is extremely rare.
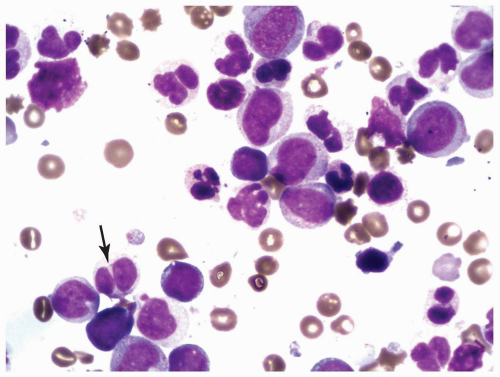 FIGURE 6.2.5 Bone marrow aspirate from a case of refractory cytopenia with multilineage dysplasia shows several hypolobated pseudo-Pelger-Huet cells (arrow). Wright-Giemsa stain, 100× magnification. |
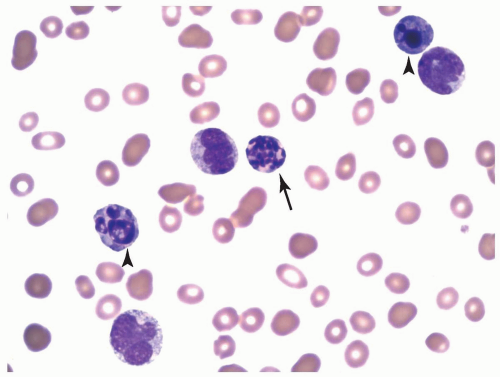 FIGURE 6.2.6 Bone marrow biopsy from a case of myelodysplastic syndrome, unclassifiable, shows a hypersegmented neutrophil (arrow) and two apoptotic cells (arrow heads). Wright-Giemsa stain, 100× magnification. |
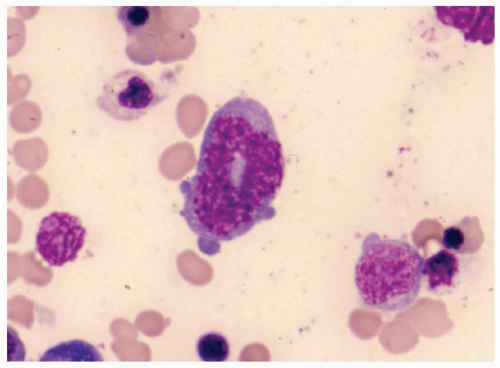 FIGURE 6.2.7 Bone marrow aspirate from a case of myelodysplastic syndrome, unclassifiable, shows a ringed nucleus in a granulocyte. Wright-Giemsa stain, 100× magnification. |
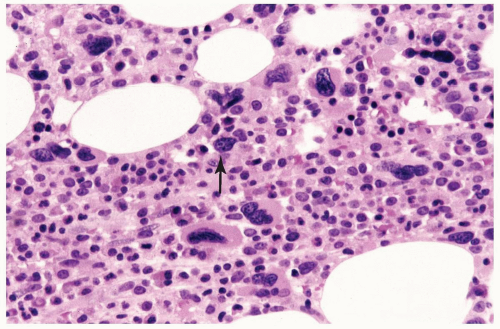 FIGURE 6.2.8 Bone marrow core biopsy from a case of refractory cytopenia with multilineage dysplasia shows many dysplastic megakaryocytes with microcytic and hypolobated morphology. Naked nuclei (arrow) are also present. Hematoxylin and eosin stain, 100× magnification. |
hypocellular MDS, which is highly responsive to immunosuppressive therapy (6).
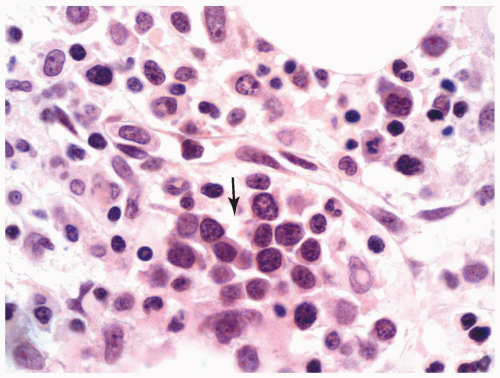 FIGURE 6.2.9 Bone marrow core biopsy from a case of refractory anemia with excess blasts shows a cluster of immature myeloid cells (arrow) in between the bony trabeculae representing abnormal localization of immature precursors (ALIP). Hematoxylin and eosin stain, 100× magnification. |
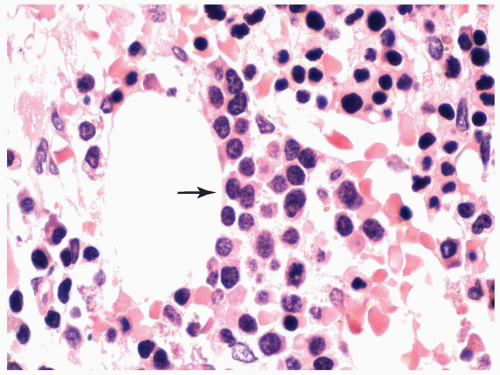 FIGURE 6.2.10 Bone marrow core biopsy from a case of refractory anemia with ringed sideroblasts shows a cluster of pronormoblasts (arrow) representing pseudo-abnormal localization of immature precursors (ALIP). The pronormoblasts have regular nuclear contour and erythroid chromatin pattern. The surrounding mature normoblasts also help to identify the erythroid origin of the immature cells. Hematoxylin and eosin stain, 100× magnification. |
is frequently present together with nucleated red blood cells in the peripheral blood. Most patients are pancytopenic with neutropenia and thrombocytopenia. Dysplastic granulocytes and atypical platelets are also present in the peripheral blood.
TABLE 6.2.2 | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
TABLE 6.2.3 | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
peripheral blood and the bone marrow (Table 6.3.1). The peripheral blood monocyte count should be >1000/µL (Fig. 6.3.2), and the monocyte count in bone marrow should be >10% (Fig. 6.3.3). Immature myeloid cells (myelocytes and metamyelocytes) can be present in the peripheral blood, but they are usually <10%. Unlike CML, CMML shows myelodysplastic changes in one or more myeloid lineages, but the Philadelphia chromosome or breakpoint cluster region/Ableson (BCR/ABL) fusion is not present. If myelodysplasia is absent or minimal, the diagnosis of CMML can still be made when peripheral monocytosis has lasted for >3 months, other causes of monocytosis have been excluded, and clonal cytogenetic abnormality is present in bone marrow cells (2). Unlike acute myeloid leukemia, CMML has <20% blasts in the blood or bone marrow.
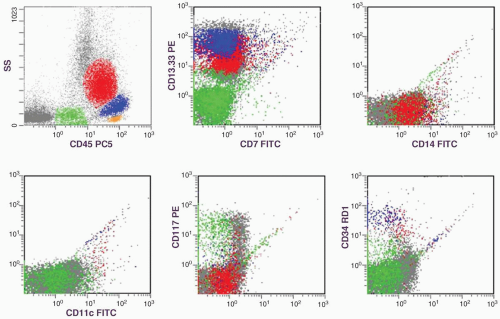 FIGURE 6.3.1 Flow cytometric histograms show three populations: the blue and red populations are mature and immature monocytes, and the green population represents immature myelocytes. The monocyte populations are positive for CD13.33, CD14, CD117, and CD34. The myelocytes are positive for CD13.33, CD117, and CD34. Both populations are negative for CD11c. SS, side scatter; PC5, phycoerythrin-cymin 5; FITC, fluorescein isothiocyanate; RD1, rhodamine. |
are classified as CMML-1. The M/E ratio in CMML is lower than that in CML and aCML, as there are >15% of erythroid components in the bone marrow (6). Mild basophilia and mild eosinophilia can be seen in some cases. If eosinophilia is striking (>1,500/µL), it becomes the variant of CMML with eosinophilia, which is associated with specific symptoms due to eosinophilic degranulation and may be associated with a specific karyotype, t(5:12) (7,8). The spleen and lymph node can be infiltrated by the myelomonocytic cells. Some patients may have generalized lymphadenopathy. These lymph nodes are characterized by extensive infiltration by plasmacytoid monocytes (9, 10, 11 and 12). The plasmacytoid monocytes have round nuclei, finely dispersed chromatin, inconspicuous nucleoli, and eosinophilic cytoplasm, but their clonal relationship with the neoplastic cells has not been proven.
TABLE 6.3.1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
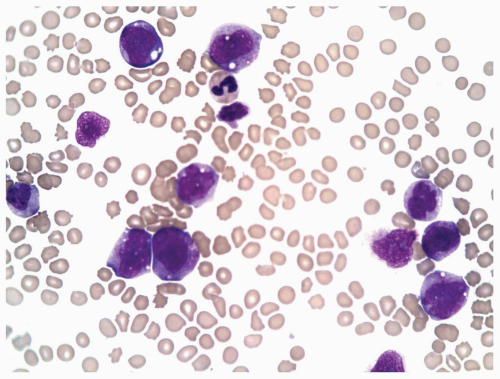 FIGURE 6.3.2 Peripheral blood smear from a case of chronic myelomonocytic leukemia (CMML)-2 shows monocytes of different developmental stages. Wright-Giemsa, 60× magnification. |
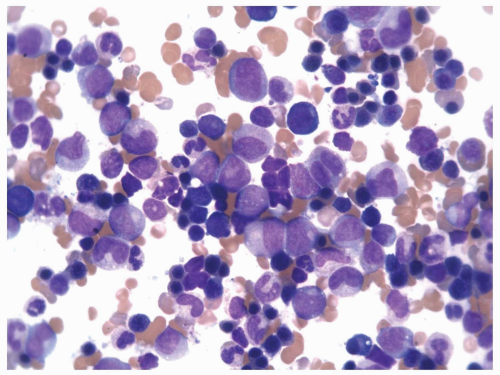 FIGURE 6.3.3 Bone marrow aspirate from a case of chronic myelomonocytic leukemia (CMML)-2 shows many mature and immature monocytes. Wright-Giemsa, 60× magnification. |
TABLE 6.3.2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
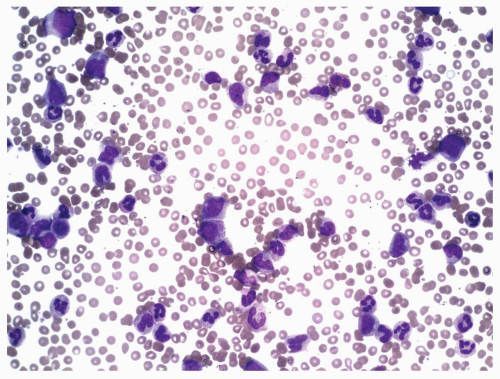 FIGURE 6.3.4 Peripheral blood smear from a case of atypical chronic myeloid leukemia (aCML) shows marked leukocytosis with several immature forms. Wright-Giemsa, 40× magnification. |
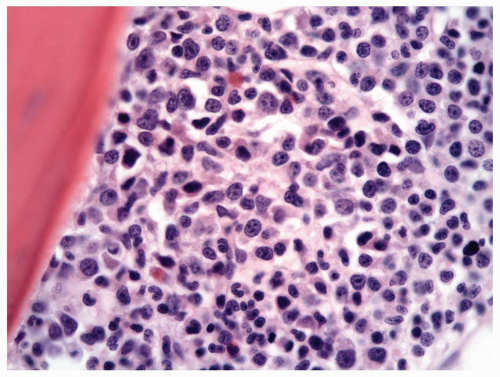 FIGURE 6.3.5 Bone marrow core biopsy shows hypercellularity with a high myeloid-erythroid (M/E) ratio. Hematoxylin and eosin, 60× magnification. |
TABLE 6.3.3 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
TABLE 6.3.4 | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
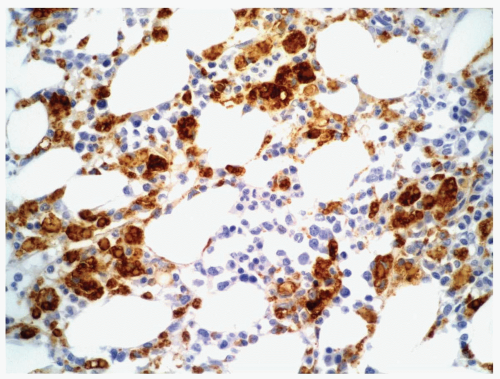 FIGURE 6.3.6 Bone marrow core biopsy from a case of chronic myelomonocytic leukemia (CMML) shows positive CD68 PG-M1 staining in many monocytes. Immunoperoxidase, 40× magnification. |
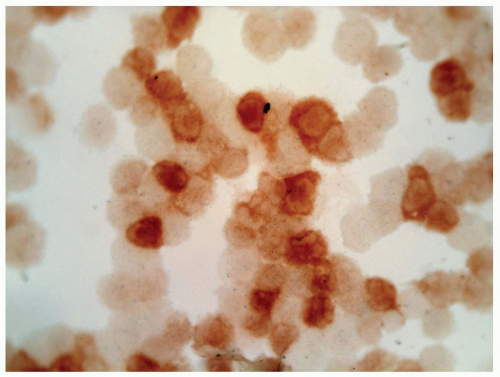 FIGURE 6.3.7 Cytospin smear of bone marrow aspirate from a patient of juvenile myelomonocytic leukemia (JMML) shows positive staining of α-naphthyl butyrate esterase in many monocytes. Cytochemical stain, 60× magnification. |
mutation in the Janus kinase 2 (JAK2) tyrosine kinase in 9 of 116 CMML/aCML cases (23). CCAAT/enhancer binding protein α (CEBPα) gene mutation is involved in CMML cases transforming into acute myeloid leukemia (24).
indicating that the absence of HLA-DR in the immunophenotype of the peripheral blood specimen was probably a technical error. The identification of this particular karyotype is clinically important because it confirms the diagnosis of AML even when the blast count in the bone marrow is <20% (1). It also confers a favorable prognosis. The presence of type 3 blasts defines the leukemia as AML-M2 irrespective of whether the mature myeloid cell count is below or above 10% (2).
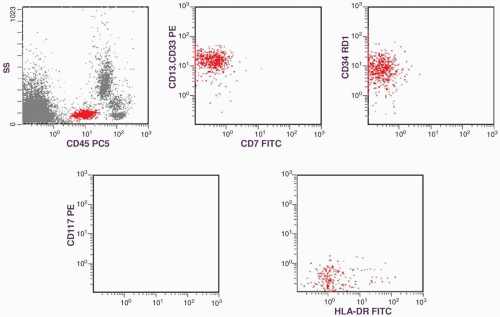 FIGURE 6.4.1 Flow cytometric analysis of bone marrow shows positive CD13.CD33, CD34, and HLA-DR, but negative CD7 and CD117. Note that a large population of mature myeloid cells is present above the gated acute myeloid leukemia (AML) population. ss, side scatter; pc, phycoerythrin-cyanin 5; PE, phycoerythrin; RD1, rhodamine; FITC, fluorescein isothiocyanate. |
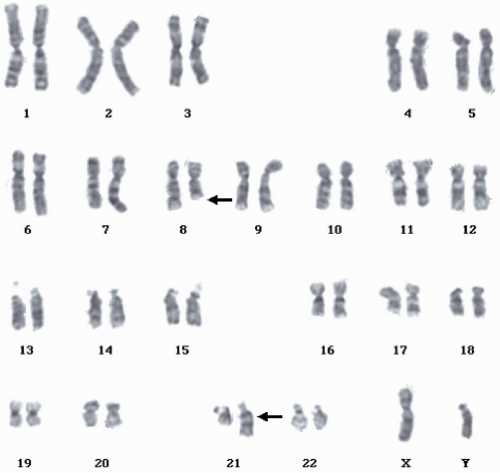 FIGURE 6.4.2 Karyotype of bone marrow reveals t(8;21)(q22;q22) (arrows). (Courtesy of Peter Papenhausen, Ph.D., LabCorp of America Cytogenetic Department, North Carolina.) |
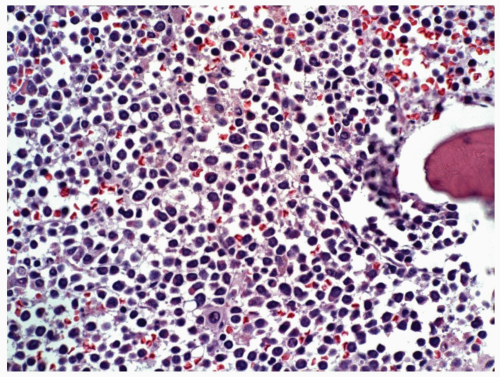 FIGURE 6.4.3 Bone marrow core biopsy shows extensive immature myeloid cell infiltration replacing normal hematopoietic cells. No normoblasts and megakaryocytes are present. Hematoxylin and eosin, 40× magnification. |
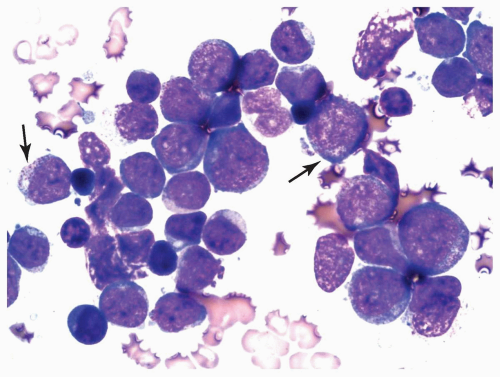 FIGURE 6.4.4 Bone marrow aspirate reveals a cluster of myeloblasts with several showing >20 cytoplasmic granules (type 3 blasts) (arrows). Wright-Giemsa, 100× magnification. |
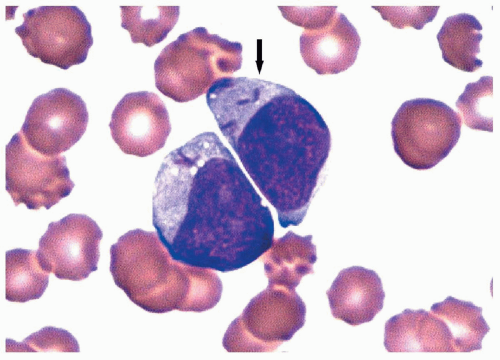 FIGURE 6.4.5 Peripheral blood smear shows two immature myeloid cells with multiple Auer rods (arrow). Wright-Giemsa, 100× magnification. |
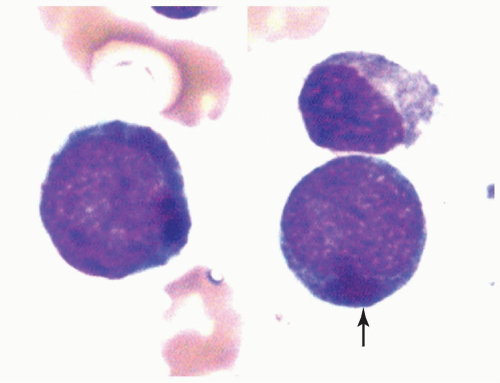 FIGURE 6.4.6 Bone marrow aspirate shows two myeloblasts containing large cytoplasmic lysosomes (pseudo-Chediak-Higashi anomaly) (arrow). Wright-Giemsa, 100× magnification. |
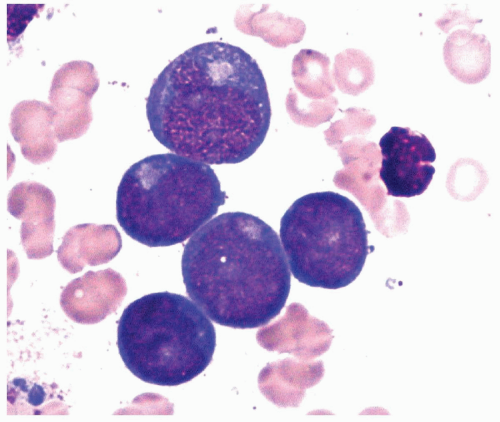 FIGURE 6.4.7 Bone marrow aspirate shows five myeloblasts with prominent Golgi. Wright-Giemsa, 200× magnification. |
t(8;21), but it was positive in only one third of the cases studied (14). In some t(8;21) cases without a positive immunohistochemical staining for PAX5, up-regulation of PAX5 transcript was identified by real-time reverse transcription-polymerase chain reaction (RT-PCR) studies (14). PAX5 is the master regulator of B-lymphopoiesis through activation of B-cell-specific genes, including CD19 and CD79a (15,16). Therefore, it is not unexpected that CD19 and CD79a are also expressed, though in lower frequency, in PAX5-positive cases. As there is no CD19 monoclonal antibody for immunohistochemistry, the identification of these two B-cell markers, PAX5 and CD79a, is most useful in surgical pathology.
TABLE 6.4.1 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
TABLE 6.4.2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
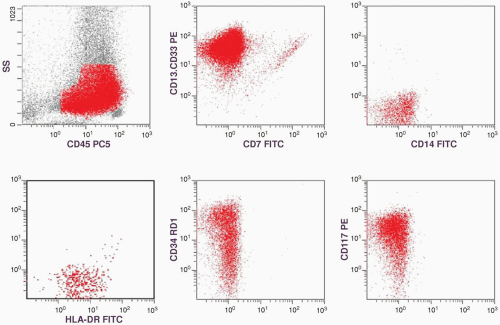 FIGURE 6.5.1 Flow cytometric analysis of bone marrow shows positive reactions with cluster of differentiation (CD)13.CD33, CD7, CD14, human leukocyte antigen-DR (HLA-DR), CD34, and CD117. ss, side scatter; PE, phycoerythrin, RD1, rhodamine; FITC, fluorescence in situ hybridization. |
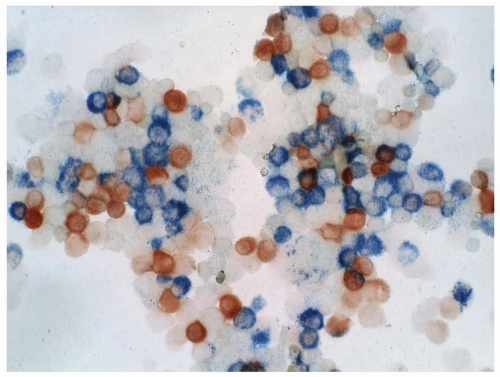 FIGURE 6.5.2 Combined esterase stain of the bone marrow cytospin shows chloroacetate esterase stain (blue) of the myeloid cells and α-naphthyl butyrate esterase stain (brown) of the monocytoid cells. 40x magnification. |
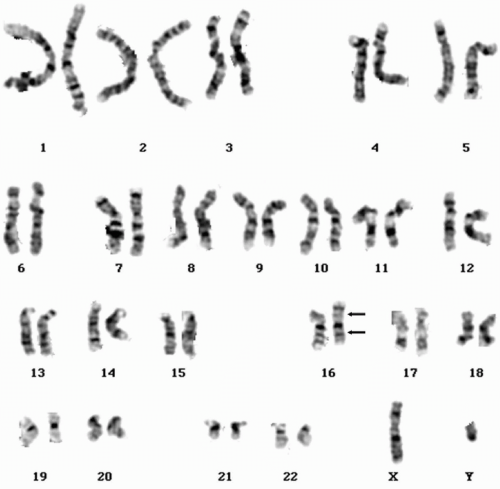 FIGURE 6.5.3 Karyotype of the bone marrow reveals inversion of chromosome 16 (arrows). (Courtesy of Peter Papenhausen, Ph.D., LabCorp of America Cytogenetics Department.) |
eosinophilia showed this karyotype (7). Inv(16) has also been reported in several cases of chronic myeloid leukemia with blast crisis, in which bone marrow eosinophilia was also observed (2).
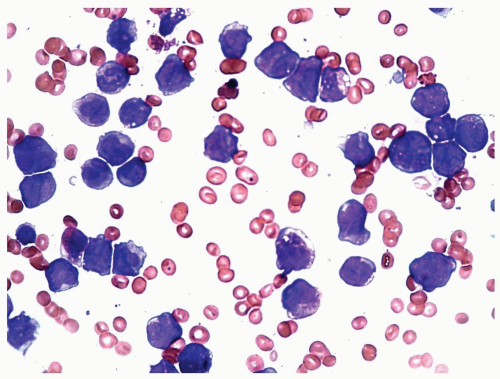 FIGURE 6.5.4 Bone marrow aspirate shows various developmental stages of myelomonocytic cells. Wright-Giemsa, 60x magnification. |
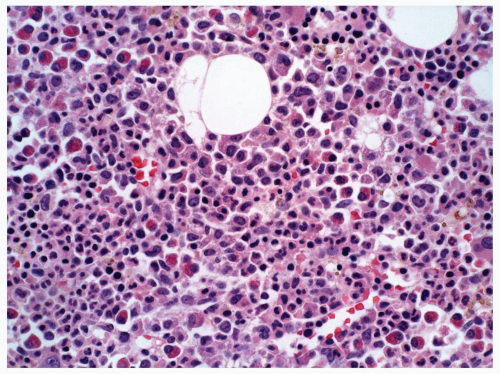 FIGURE 6.5.5 Bone marrow core biopsy reveals hypercellular marrow composed of immature myelomonocytic cells. Eosinophilia is evident. Hematoxylin and eosin, 40x magnification. |
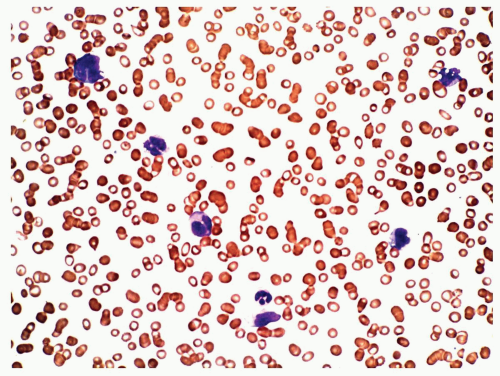 FIGURE 6.5.6 Peripheral blood smear shows monocytosis. Wright-Giemsa, 60x magnification. |
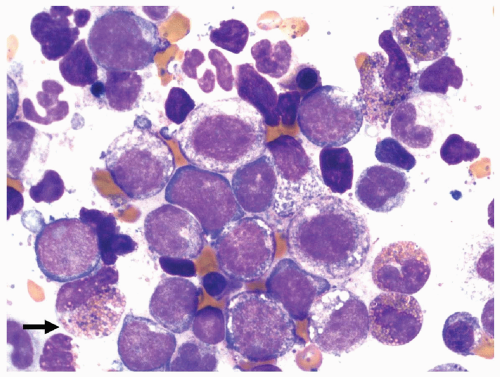 FIGURE 6.5.7 Bone marrow aspirate reveals immature myelomonocytic cells with the presence of basophilic granules in a few immature eosinophils (arrow). 100x magnification. |
TABLE 6.5.1 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
 FIGURE 6.6.1 Bone marrow core biopsy shows total replacement of normal hematopoietic cells by the leukemic promyelocytes. Note many bilobed cells (arrow) are present. The abundant eosinophilic cytoplasm represents hypergranularity. Hematoxylin and eosin, 100x magnification. |
 FIGURE 6.6.2 Flow cytometric histograms show strongly positive CD33 and weakly positive CD34 but negative human leukocyte antigen-DR (HLA-DR). ss, side scatter; PE, phycoerythrin; FITC, fluorescence in situ hybridization; RD1, rhodamine; PC5, phycoerythrin cyanin 5. |
 FIGURE 6.6.3 Combined esterase stain of the bone marrow aspirate shows multiple Auer rods demonstrated by chloroacetate esterase (blue) stain in a few leukemic cells (arrow). 100x magnification. |
 FIGURE 6.6.4 Peripheral blood smear shows several hypergranular promyelocytes with the nuclei being masked by the cytoplasmic granules. Note one promyelocyte contains multiple Auer rods (arrow). Wright-Giemsa, 100x magnification. |
 FIGURE 6.6.5 Bone marrow aspirate shows many leukemic promyelocytes, and two reveal multiple Auer rods (arrows). Wright-Giemsa, 100x magnification. |
granules, and conspicuous cytoplasmic budding mimicking micromegakaryocytes (8,9). A hand-mirror variant of M3v has also been described (10).
 FIGURE 6.6.6 Peripheral blood smear shows several hypogranular promyelocytes with bilobed or folded nuclei, mimicking monocytes. Wright-Giemsa, 100x magnification. |
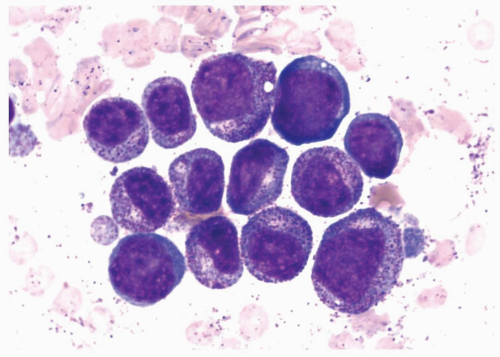 FIGURE 6.6.7 Bone marrow aspirate shows a cluster of hypergranular promyelocytes due to synchronous proliferation of promyelocytes after chemotherapy of M2 leukemia. Wright-Giemsa, 100x magnification. |
that this phenomenon might be related to a non-European genetic factor.
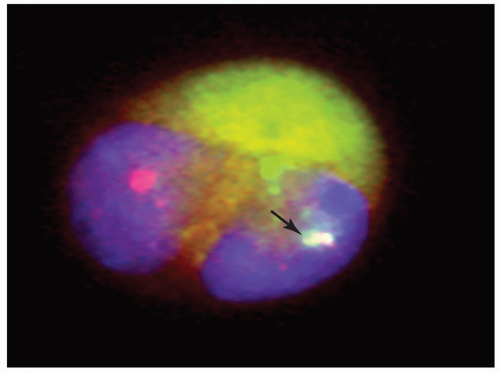 FIGURE 6.6.8 Fluorescence in situ hybridization of bone marrow aspirate with promyelocytic leukemia-retinoic acid receptor α (PML-RARα) probes demonstrated one orange signal, one green signal, and the PML-RARα fusion product (arrow) in a leukemic promyelocyte with a bilobed nucleus. 100x original magnification. |
TABLE 6.6.1 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
informed of the diagnosis of acute myeloid leukemia (AML), the prognosis, and treatment of the disease. He decided to forego chemotherapy and seek possible palliative care at home. The patient was discharged with the instruction to follow up by visiting hematology/oncology, and cardiology clinics.
 FIGURE 6.7.1 Flow cytometric histograms of the bone marrow show dual CD13-CD33/CD7 staining, with positive HLA-DR, CD117, and CD34. ss, side scatter; PC5, phycoerythrin cyanin 5; FITC, fluorescein isothiocyanate; RD1, rhodamine; PE, phycoerythrin. |
varies from 2% to 22% in different series (5, 6, 7, 8 and 9). M0 is frequently associated with the presence of terminal deoxynucleotidyl transferase (TdT) (9).
TABLE 6.7.1 | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
 FIGURE 6.7.2 Myeloperoxidase stain of the bone marrow shows positive staining in several myeloblasts as well as maturing myeloid cells. 100x magnification. |
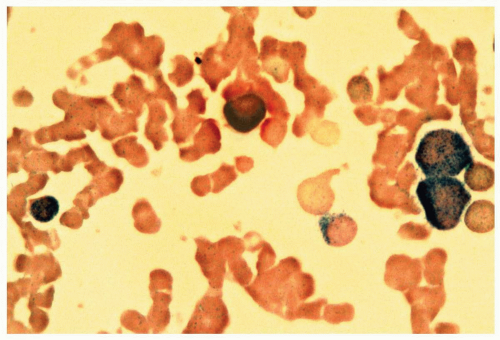 FIGURE 6.7.3 Combined esterase stain reveals chloroacetate esterase staining (blue) in two myeloblasts and one granulocyte, and α-naphthyl butyrate staining (brown) in a monocyte. 100x magnification. |
TABLE 6.7.2 | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
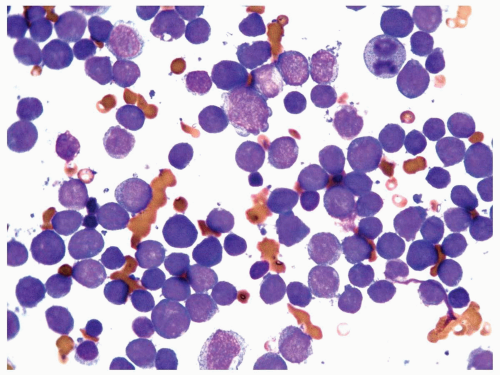 FIGURE 6.7.4 Bone marrow aspirate reveals almost exclusively blasts in the marrow with a high nuclear/cytoplasmic ratio, immature chromatin pattern, and nucleoli. 60x magnification. |
of CD13-CD33 is low but that of MPO is high. That is why both of these myeloid markers should be included in flow cytometric studies of AML. The presence of high percentages of CD34- and CD117-positive cells and the dual CD13-CD33/CD7 staining are supportive of a malignant cell population, which will be discussed in the immunophenotype section. With all this information, a diagnosis of AML without maturation (M1) was established.
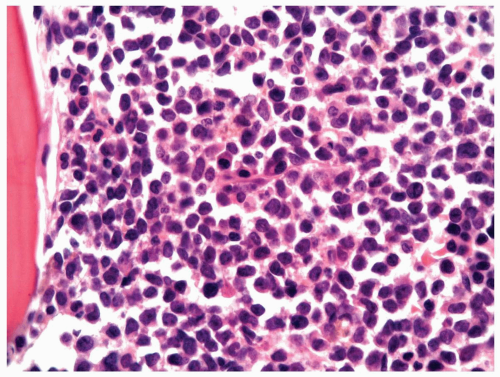 FIGURE 6.7.5 Bone marrow aspirate shows 100% cellularity, and normal hematopoietic cells are almost totally replaced by blastic cells. 60x magnification. |
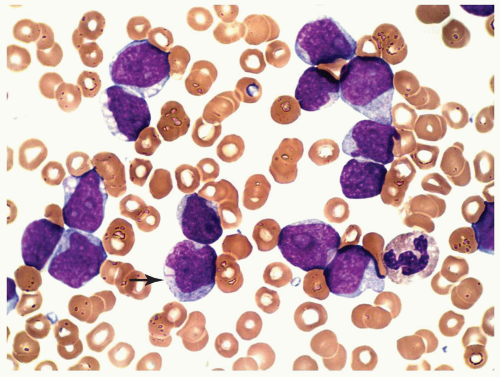 FIGURE 6.7.6 Peripheral blood smear shows multiple myeloblasts with similar features as those seen in the bone marrow. One Auer rod in the cytoplasm of a blast is indicated (arrow). 100x magnification. |
disorders. Although CD34 may also be detected in cases of MDS, its percentage is usually lower than that in AML cases. When a high percentage is present in a case of MDS, it predicts leukemic transformation (26). CD34 is found more frequently in M0, M1, and M5a (27,28) but is often absent in M3 (29). It is associated with either good or poor prognosis, depending on the cytogenetic alteration in a particular case and the cell lineage (25). Generally, it predicts poor prognosis in AML but good prognosis in ALL (28). When CD34 is related to poor prognosis, it is usually due to the correlation between CD34 and the multiple drug resistance (MDR) protein (25). CD34 may appear at relapse of CD34-negative AML, supporting its being an unfavorable marker (28).
TABLE 6.7.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 6.7.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
to contribute to disease progression. Primary aberrations often involve structural changes (e.g., reciprocal translocations and inversions), whereas the secondary aberrations usually involve genomic imbalances (trisomies, monosomies, deletions, and unbalanced translocations).
TABLE 6.7.5 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
CD34 positivity, and elevated lactate dehydrogenase levels (3,61). The identification of FLT3 mutations and overexpression of ecotropic viral integration site 1 (EVI 1) have been included recently as independent indicators for an unfavorable prognosis (14). In general, AML carries a worse prognosis than ALL. In adult AML cases, the cure rates are approximately 40% to 50%, as compared to the 75% to 80% cure rates in pediatric ALL cases (62).
marker: CD7 0%. Immature cell markers: CD34 51%, CD117 74% (Fig. 6.8.1).
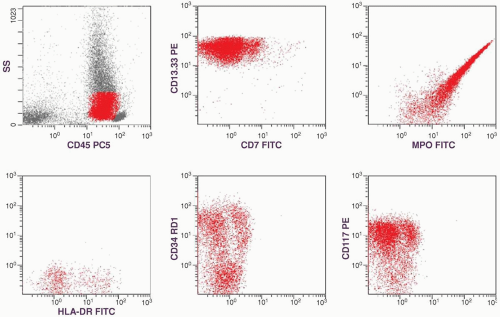 FIGURE 6.8.1 Flow cytometric analysis of bone marrow shows a tight cluster of leukemic cells in the cluster of differentiation (CD)45/side-scatter gating and a large population of maturing granulocytes with high side scatter. The population of leukemic cells is characterized by myeloid markers CD13-CD33, myeloperoxidase (MPO), and by stem cell markers CD34 and CD117. Human leukocyte antigen-DR (HLA-DR) is weekly positive. ss, side scatter; PC5, phycoerythrin cyanin 5; PE, phycoerythrin; FITC, fluorescein isothiocyanate; RD1, rhodamine. |
marrow (Figs. 6.8.3 and 6.8.4). No monoblasts were demonstrated in the bone marrow, and <1% of monocytes were present. Therefore, morphologically, it fulfils the definition of AML with maturation. However, if monoblasts are present and the distinction between the myeloblast and monoblast populations is not certain, cytochemical staining is required to distinguish M2 from M4.
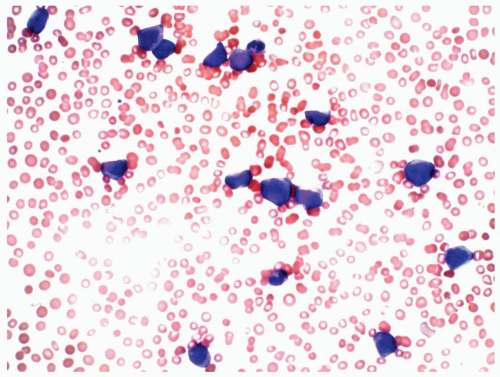 FIGURE 6.8.2 Peripheral blood smear shows myeloblasts and a few mature granulocytes. Wright-Giemsa, 40x magnification. |
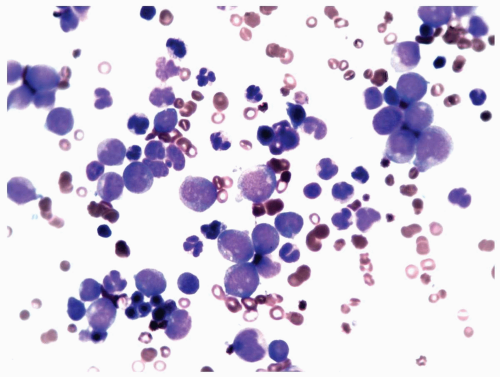 FIGURE 6.8.3 Bone marrow aspirate reveals a high percentage of myeloblasts intermingled with mature granulocytes. Wright-Giemsa, 60x magnification. |
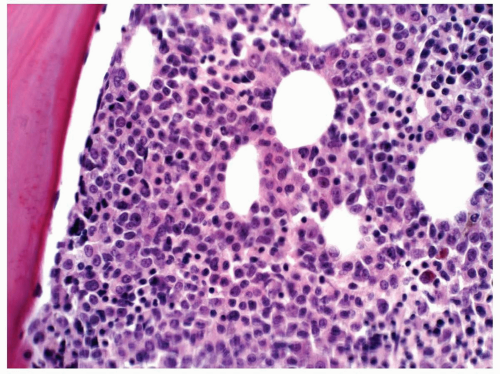 FIGURE 6.8.4 Bone marrow core biopsy demonstrates hypercellularity with mature and immature myeloid cells. No erythroid elements are demonstrated. Hematoxylin and eosin, 40x magnification. |
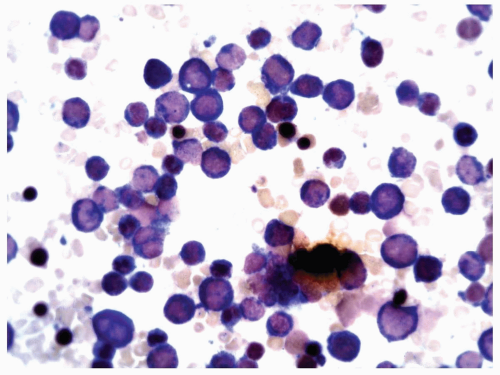 FIGURE 6.8.5 Bone marrow aspirate stained with combined esterases. All myeloblasts and different developmental stages of myeloid cells stained with chloroacetate esterase (blue). Only one monocyte is stained by α-naphthyl butyrate esterase (brown). Combined esterase stain, 60x magnification. |
 FIGURE 6.8.6 Bone marrow biopsy shows scattered CD34-positive myeloblasts. Immunoperoxidase, 40x magnification. |
different from other subtypes of AML. The major mechanisms are failure of the leukemic cells to mature and inhibition of normal hematopoiesis. As a result, the patients may have anemia, neutropenia, and/or thrombocytopenia. This subtype of AML usually responds well to chemotherapy, but the prognosis is frequently associated with the abnormal karyotype of the leukemic cells.
TABLE 6.8.1 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
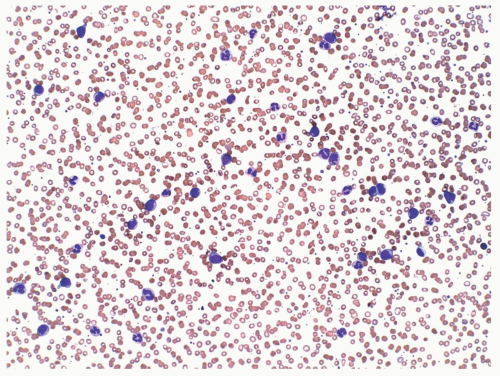 FIGURE 6.9.1 Peripheral blood smear shows leukocytosis consisting of mature and immature myelomonocytic cells. Wright-Giemsa, 20x magnification. |
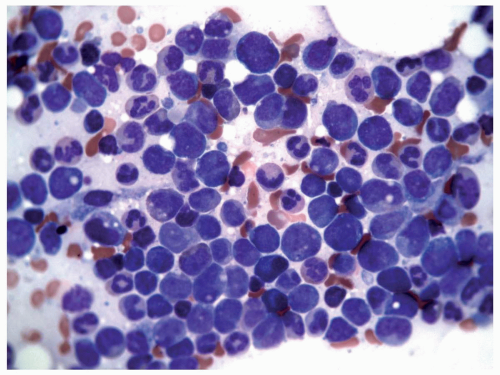 FIGURE 6.9.2 Bone marrow aspirate shows a packed marrow with mostly immature myeloid and monocytoid cells. Wright-Giemsa, 60x magnification. |
particularly difficult by morphology. However, nonspecific esterase can be weak or absent in monocytoid cells in some cases. If morphologic identification of monocytes is certain, absence of nonspecific esterase does not exclude the diagnosis of M4 (2).
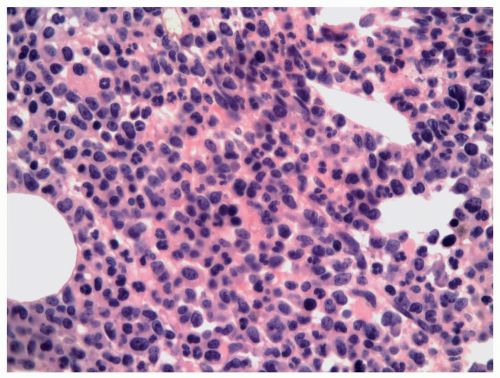 FIGURE 6.9.3 Bone marrow core biopsy reveals hypercellular bone marrow with myelomonocytic leukemic cells replacing the normal hematopoietic components. Hematoxylin and eosin, 20x magnification. |
 FIGURE 6.9.4 Flow cytometric histograms from a case of acute myelomonocytic leukemia (not the current case) demonstrated two immature cell clusters in the dot plot. Both populations express CD117, CD13-CD33, and (partially) CD7. The red cluster also shows CD14 and human leukocyte antigen-DR (HLA-DR). PC5, phycoerythrin cyanin 5; FITC, fluorescein isothiocyanate; PE, phycoerythrin; SS, side scatter. |
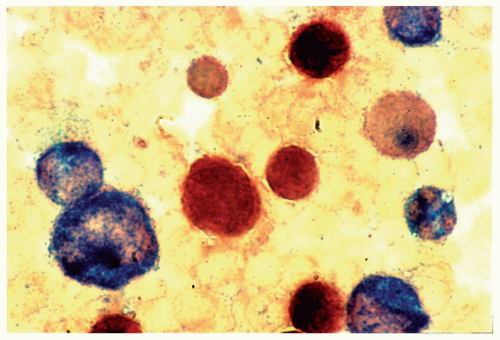 FIGURE 6.9.5 A bone marrow aspirate reveals chloroacetate esterase-positive (blue) and α-naphthyl butyrate esterase-positive (brown) populations. Note that some cells are positive for both esterases. Combined esterase stain, 100x magnification. (From Sun T. Flow cytometric analysis of hematologic neoplasms, 2002.) |
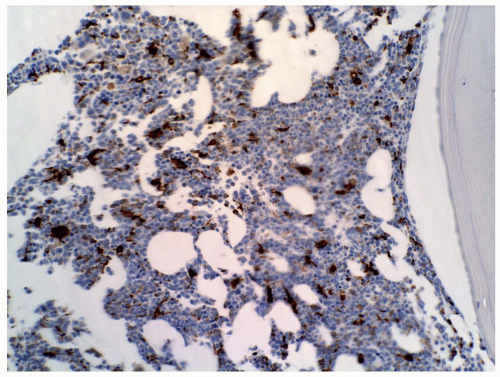 FIGURE 6.9.6 Bone marrow core biopsy shows many cells with CD68 PG-M1 staining. Myeloperoxidase stain, 20x magnification. |
TABLE 6.9.1 | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
tumor lysis. The patient was discharged and died at home on the day of discharge.
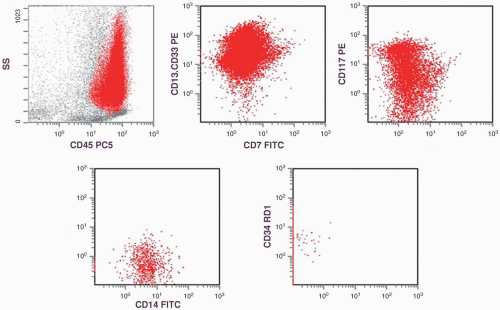 FIGURE 6.10.1 Flow cytometric analysis of bone marrow in an M5 case shows positive reactions to cluster of differentiation (CD)7, CD13.CD33, CD14, and CD117, but negative reactions to CD34. These histograms are not from the current case. SS, side scatter; PE, phycoerythrin; PC5, phycoerythrin cyanin 5; FITC, fluorescein isothiocyanate; RD1, rodamine. |
origin on the short arm of the other chromosome 16 in addition to the t(8;16).
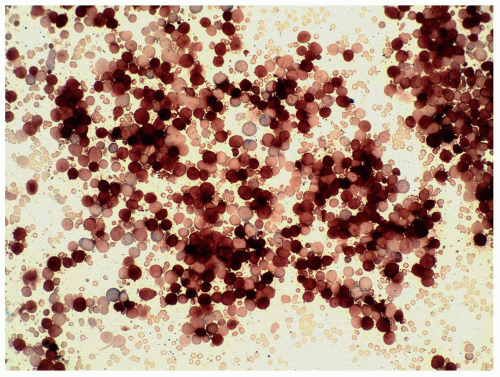 FIGURE 6.10.2 Combined esterase stain of a bone marrow aspirate shows predominantly nonspecific esterase staining of the blasts. Specific esterase stains only a few mature myeloid cells. 40x magnification. |
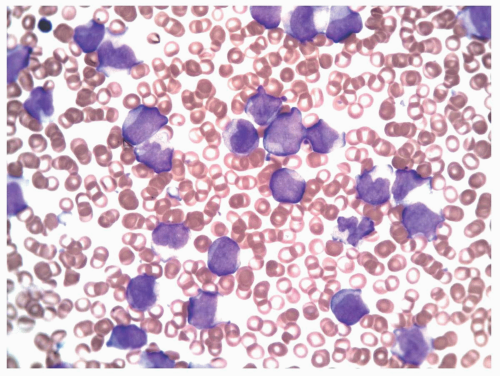 FIGURE 6.10.3 Peripheral blood smear of an M5 case shows various monocytic stages. Wright-Giemsa, 60x magnification. |
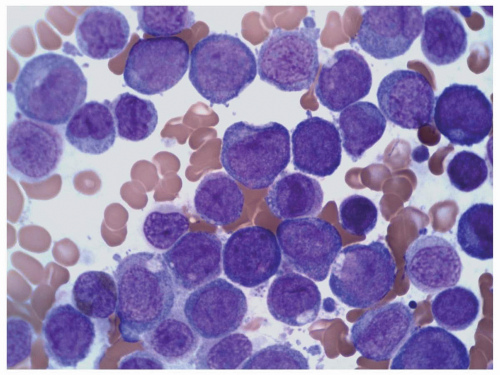 FIGURE 6.10.4 Bone marrow aspirate of an M5 case shows many monoblasts and promonocytes with a few monocytes. Wright-Giemsa, 100x magnification. |
and immunophenotyping is necessary for an accurate diagnosis of M5.
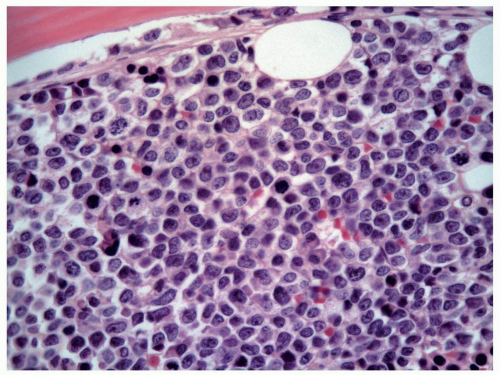 FIGURE 6.10.5 Bone marrow biopsy of an M5 case shows various stages of monocytes, replacing the normal hematopoietic cells. Hematoxylin and eosin, 60x magnification. |
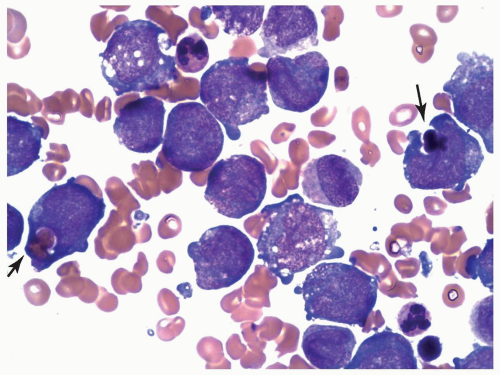 FIGURE 6.10.6 Bone marrow aspirate of an M5 case with t(8;16) shows phagocytosis of erythrocytes and normoblasts by monoblasts (arrows). Wright-Giemsa, 100x magnification. |
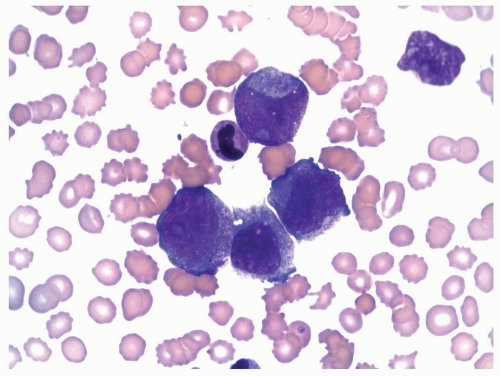 FIGURE 6.10.7 Peripheral blood smear of an M5 case with t(8;16) shows hypergranular monoblasts. Wright-Giemsa, 100x magnification. |
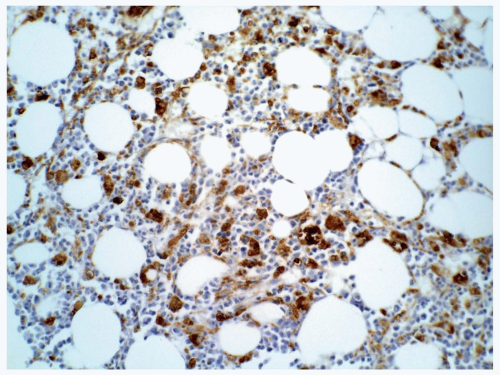 FIGURE 6.10.8 Bone marrow biopsy of an M5 case shows extensive staining of CD68 PG-M1. Immunoperoxidase, 20x magnification. |
median survival of 1 and 3 weeks, respectively, whereas patients with no renal failure or low B2M levels have a median survival of 26 and 29 weeks, respectively. In the same study, lysozyme, lactate dehydrogenase, and B2M levels were elevated in 88%, 68%, and 81% of M5 cases, respectively, but the first two components did not show statistically significant association with patient prognosis (3).
TABLE 6.10.1 | ||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||
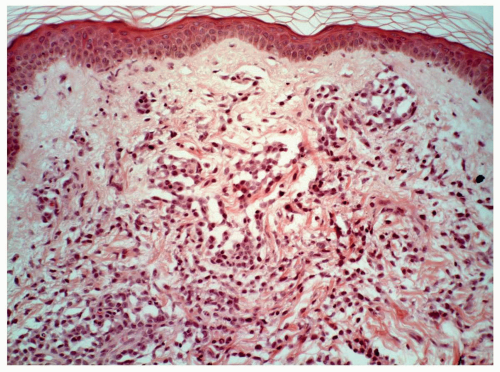 FIGURE 6.10.9 Skin biopsy of an M5 case shows extensive leukemic cell infiltration in the dermis. Hematoxylin and eosin, 20x magnification. |
cells, which included no myeloblasts. The pronormoblasts were pleomorphic with multiple intraplasmic vacuoles that were positive for periodic acid-Schiff (PAS) stain. The maturing normoblasts revealed megaloblastoid changes and nuclear dysplasia.
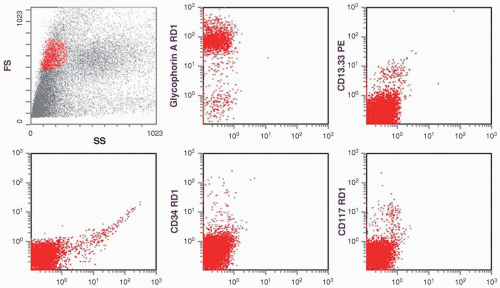 FIGURE 6.11.1 Flow cytometric analysis using the forward scatter (FS) versus side scatter (SS) plot, as the normoblasts are CD45 dim or negative. The histograms show positive glycophorin A reaction, but negative reactions for human leukocyte antigen-DR (HLA-DR), CD34, and CD117. A low percentage of CD13-CD33 is detected. FITC, fluorescein isothiocyanate; RD1, rhodamine; PE, phycoerythrin. |
erythroid leukemia as AML-M6 variant. Mazzella et al. (7) further subdivided AMA-M6 into M6a, M6b, and M6c (7). This group defined M6c cases as those with both myeloblasts and pronormoblasts >30% of the nucleated cells.
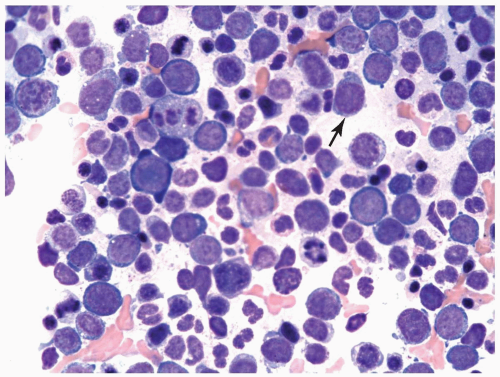 FIGURE 6.11.2 Bone marrow aspirate from a case of erythroleukemia (erythroid/myeloid) shows mainly pronormoblasts with a few myeloblasts (arrow). Wright-Giemsa, 60× magnification. |
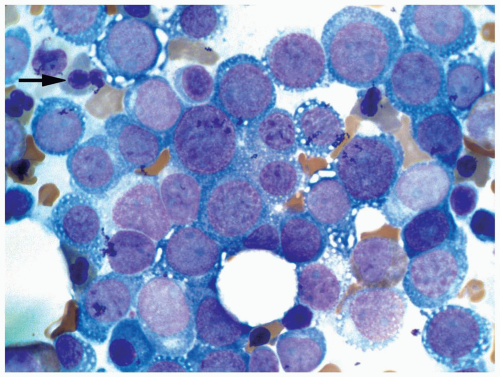 FIGURE 6.11.3 Bone marrow aspirate from a case of pure erythroid leukemia reveals predominantly pronormoblasts with other stages of normoblasts. Myeloblasts are absent. Note cytoplasmic vacuolation in pronormoblasts and a few dysplastic nucleated erythrocytes (arrow). Wright-Giemsa, 100× magnification. |
stains are myeloblasts (Fig. 6.11.5) (14). Some early normoblasts may show weak focal α-naphthyl butyrate esterase stain, which is, however, not helpful in the diagnosis. The Prussian blue stain for iron is helpful to detect ringed sideroblasts, which are seen more frequently in M6b than in M6a cases (2).
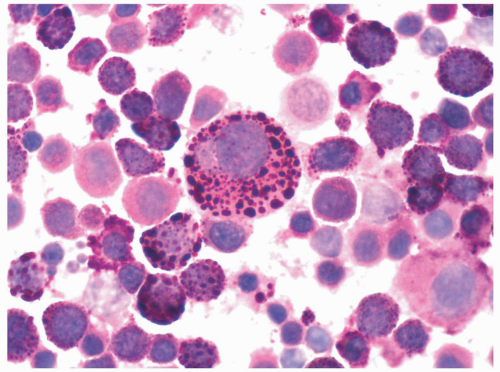 FIGURE 6.11.4 Bone marrow aspirate from a case of pure erythroid leukemia shows coarse periodic acid-Schiff (PAS)-positive granules in the cytoplasm of pronormoblasts and basophilic normoblasts, but diffuse staining in polychromatophilic and orthochromatic normoblasts. PAS, 100× magnification. |
 FIGURE 6.11.5 Bone marrow aspirate from a case of M6a reveals myeloperoxidase staining in the myelocytic series including myeloblasts, but the normoblasts are negative. Myeloperoxidase, 100× magnification. |
 FIGURE 6.11.6 Bone marrow core biopsy from a case of erythroid leukemia shows normal hematopoietic cells are replaced by erythrocytic series. Hematoxylin and eosin, 40× magnification. |
 FIGURE 6.11.7 Pronormoblasts in a bone marrow biopsy readily recognizable in a Giemsa-stained preparation. Giemsa, 100× magnification. |
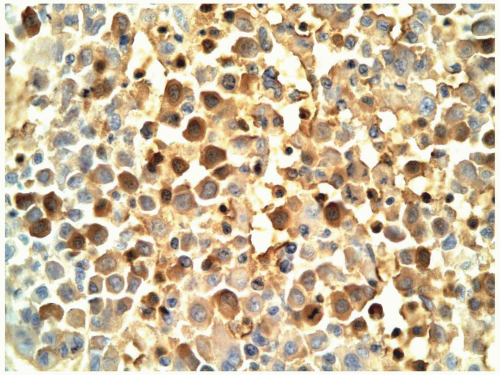 FIGURE 6.11.8 Erythroid cells in an M6 case are highlighted by hemoglobin A staining. 60× magnification. |
stem cell. Multidrug resistance gene expression and p53 gene mutation were demonstrated in all subtypes of M6, but more frequently in M6b and M6c than in M6a (2).
TABLE 6.11.1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
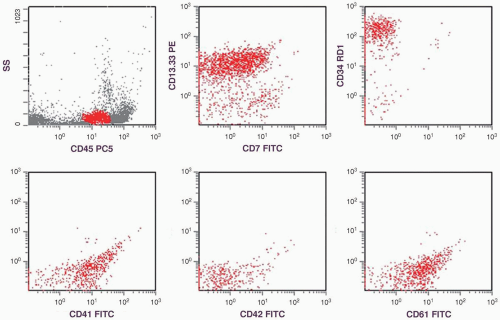 FIGURE 6.12.1 Flow cytometric analysis shows positive CD13.CD33, partially positive CD7, and positive CD34 reactions. The diagnostic feature is the expression of the immature megakaryocyte markers (CD41 and CD61) with low percentage or negative reaction of the mature megakaryocytic marker (CD42). SS, side scatter; PC5, phycoerythrin cyanin 5; PE, phycoerythrin; RD1, rhodamine; FITC, fluorescein isothiocyanate. |
Under this condition, unequivocal megakaryoblasts should be identified in the peripheral blood and/or bone marrow by immunologic techniques (7).
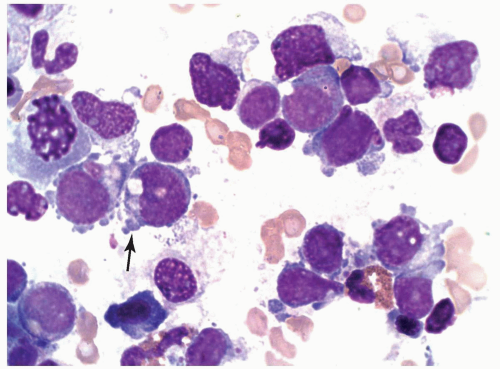 FIGURE 6.12.2 Bone marrow aspirate shows a cluster of megakaryoblasts of varying sizes. Large cells show moderate amount of basophilic cytoplasm with irregular cell border or budding (arrow). Chromatin is dispersed and nucleoli are present. Small cells, in contrast, reveal scanty cytoplasm and dense chromatin, resembling lymphoblasts. Wright-Giemsa, 100× magnification. |
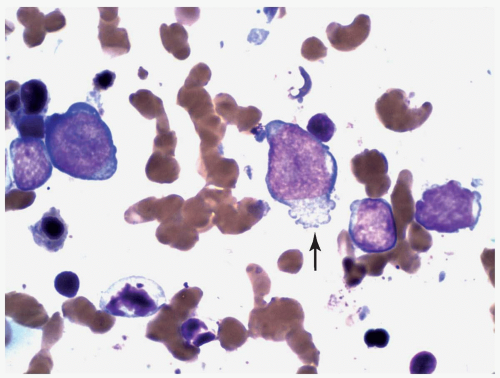 FIGURE 6.12.3 Bone marrow touch imprint shows two large and two small megakaryoblasts. Cytoplasmic projection is clearly visible in the large blasts (arrow). Wright-Giemsa, 100× magnification. |
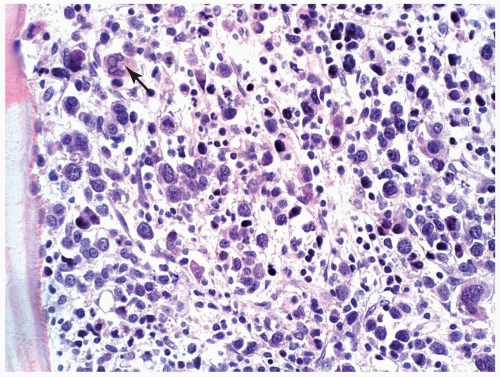 FIGURE 6.12.4 Bone marrow biopsy shows total replacement of the normal hematopoietic cells by megakaryoblasts and a few large megakaryocytes (arrow). Marked variation in size of the blasts distinguishes megakaryoblasts from other blasts. Hematoxylin and eosin, 40× magnification. |
7 to 10 µm in diameter, hypolobated, or mononucleated (11). These megakaryocytes are often referred to as micromegakaryocytes or dwarf megakaryocytes. They may be present singly or forming small or large clusters. A reticulin fiber network usually surrounds the mature megakaryocytes, but prominent fibrosis may not be present, especially in cases where immature megakaryoblasts are predominant.
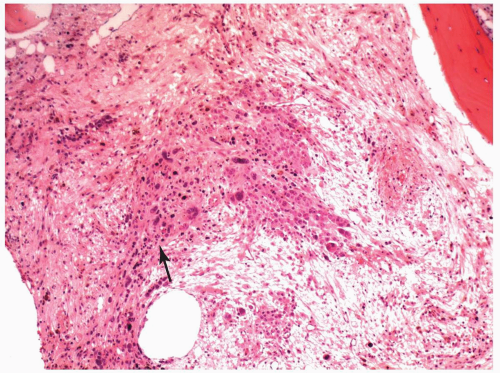 FIGURE 6.12.5 Bone marrow biopsy in a case of M7 with relapse shows a cluster of leukemic cells (arrow) on a fibrotic background. Hematoxylin and eosin, 10× magnification. |
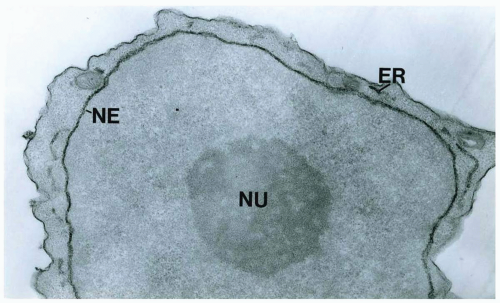 FIGURE 6.12.6 Electron micrograph of a megakaryoblast in a case of M7 leukemia, showing platelet peroxidase activity in the endoplasmic reticulum (ER) including the nuclear envelope (NE). Note the prominent nucleolus (NU) in the nucleus. 30,000× magnification. (Courtesy of Dr. Saul Teichberg, North Shore University Hospital, New York.) |
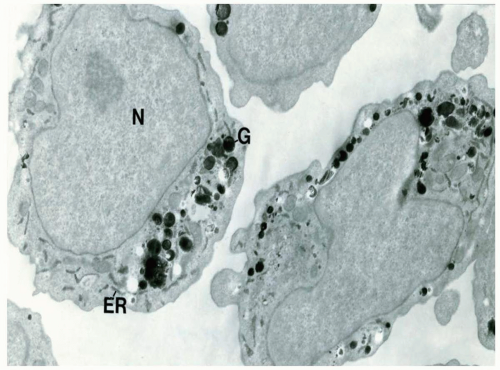 FIGURE 6.12.7 Electron micrograph of a myeloblast showing myeloperoxidase activity in many secretory granules (G) but also in the endoplasmic reticulum (ER) including the nuclear envelope. Note the immature nucleus (N) with a prominent nucleolus in one cell. 11,500× magnification. (Courtesy of Dr. Saul Teichberg, North Shore University Hospital, New York.) |
(1, 2 and 3,8,13,20). Recently, CD36 has been included in the megakaryocytic profile (4,9). In paraffin sections, factor VIII antibody was frequently used to identify megakaryocytes, but factor VIII may not be present on the leukemic cells in all patients with AMKL (12). Currently, CD42b and CD61 antibodies are available for immunohistochemical staining and provide a more specific identification (9).
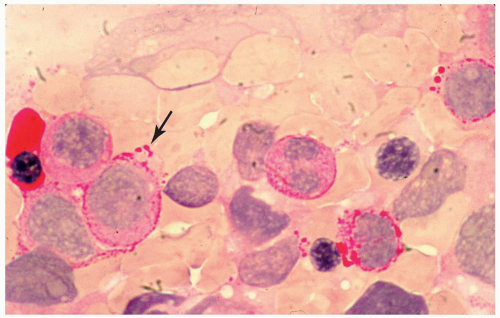 FIGURE 6.12.8 Bone marrow aspirate with periodic acid-Schiff stain shows a peripheral staining pattern with accentuation in cytoplasmic blebs (arrow). 100× magnification. |
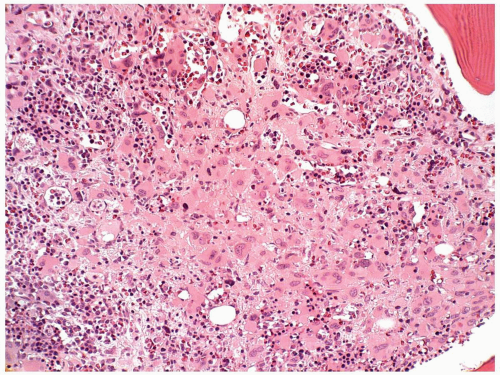 FIGURE 6.12.9 Bone marrow biopsy from a patient with acute myeloblastic leukemia shows extensive megakaryocytic reaction. Hematoxylin and eosin, 20× magnification. |
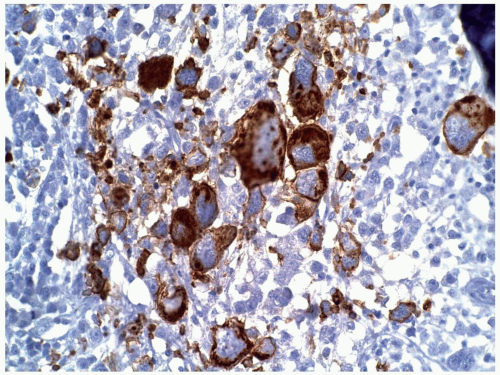 FIGURE 6.12.10 Bone marrow biopsy with CD42b staining shows positive stain in the relatively mature megakaryocytes/megakaryoblasts. Myeloperoxidase, 40× magnification. |
TABLE 6.1 2.1 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
(1,42). However, pediatric patients with t(1;22) may often have organomegaly (34). Patients with AMKL are usually anemic and thrombocytopenic. The leukocyte count may be low at the beginning of the disease, but an abrupt and rapid increase in the number of peripheral blasts is frequently seen in the terminal stage. The platelet aggregation responses may be impaired, and serum lactate dehydrogenase levels are frequently elevated. Bilateral symmetrical periostitis and osteolytic lesions have been observed in children (43).
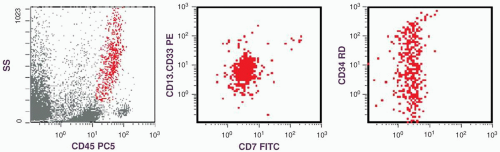 FIGURE 6.13.1 Flow cytometric analysis of a lymph node shows a large cluster of granulocytic cells (red) in the dot plot. Gated cell cluster shows a dual CD7 and CD13.CD33 staining as well as a spectrum of CD34 staining, representing mature and immature granulocytes. SS, side scatter; PC5, phycoerythrin cyanin 5; PE, phycoerythrin; FITC, fluorescein isothiocyanate. |
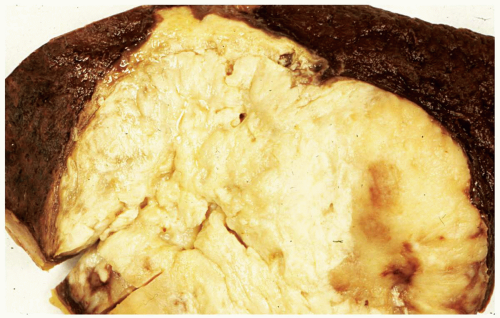 FIGURE 6.13.2 Splenectomy specimen shows a large solid tumor mass in the center, representing a granulocytic sarcoma. |
in size and show nuclear grooves, creases, or convolutions (7). The cytoplasm is moderate to abundant, and a small number of tumor cells may show cytoplasmic granules consistent with myeloid differentiation. The blastic group is formed predominantly of myeloblasts. In tissue sections, the nuclei of the tumor cells are uniform and relatively round. The nuclear chromatin is dispersed, and small inconspicuous nucleoli are seen only occasionally. The cytoplasm varies in amount and contains no granules.
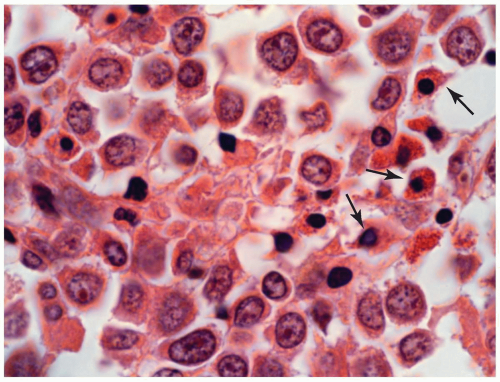 FIGURE 6.13.3 Myeloid sarcoma of bone shows a few eosinophilic myelocytes (arrows) scattered among the large tumor cells. Hematoxylin and eosin, 100× magnification. |
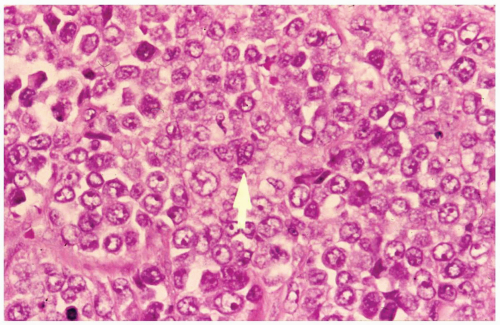 FIGURE 6.13.4 Myeloid sarcoma of the hip shows large tumor cells with vesicular nuclei and conspicuous nucleoli. One cell shows a nuclear groove (arrow). Hematoxylin and eosin, 60× magnification. |
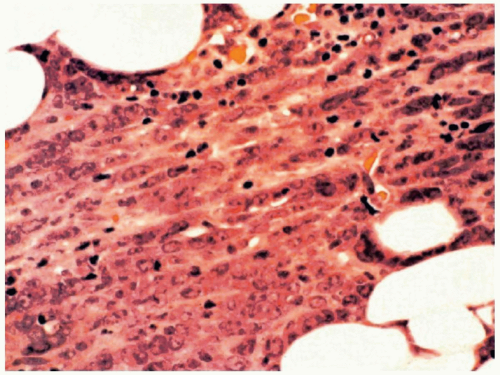 FIGURE 6.13.5 Myeloid sarcoma of a lymph node shows a cording pattern at the periphery of the tumor. Hematoxylin and eosin, 40× magnification. |
carcinomas (11). Electron microscopy may help in the differential diagnosis by demonstrating specific cytoplasmic granules and/or Auer rods, but the most useful technique for a definitive diagnosis is immunophenotyping by immunohistochemistry or flow cytometry.
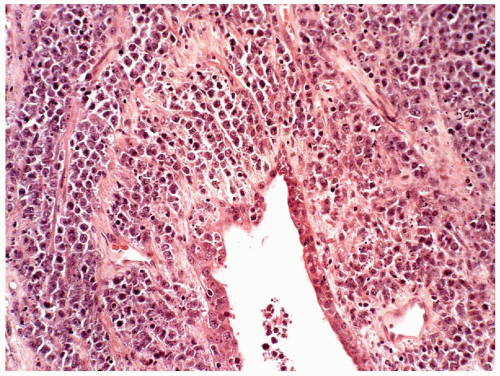 FIGURE 6.13.6 Myeloid sarcoma of the lung shows cords of tumor cells around the bronchus. Hematoxylin and eosin, 20× magnification. |
TABLE 6.13.1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
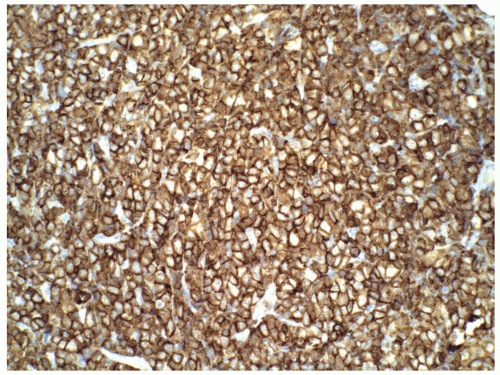 FIGURE 6.13.7 Myeloid sarcoma cells stain strongly with CD43. Immunoperoxidase, 20× magnification. |
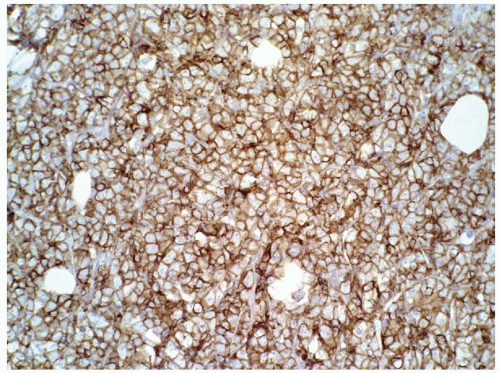 FIGURE 6.13.8 Myeloid sarcoma cells stains strongly with CD45, but negative with CD20 and CD3 (not shown). Immunoperoxidase, 20× magnification. |
TABLE 6.1 3.2 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
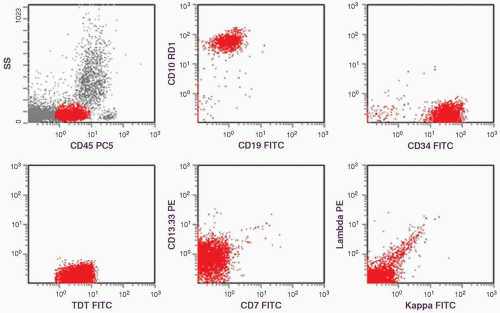 FIGURE 6.14.1 Flow cytometric histograms show dual staining of CD10 and CD19, positive CD34 and terminal deoxynucleotidyl transferase (TdT), partial positive CD13.33, but negative CD7, kappa, and lambda. SS, side scatter; PC5, phycoerythrin-cyanin 5; RD1, rhodamine; FITC, fluorescein isothiocyanate; TDT, terminal deoxynucleotidyl transferase; PE, phycoerythrin. |
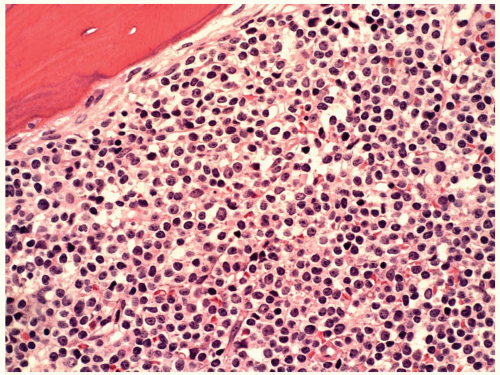 FIGURE 6.14.2 Bone marrow biopsy of a case of B-cell acute lymphoblastic leukemia (ALL) shows diffuse blast infiltration. Hematoxylin and eosin, 40× magnification. |
TABLE 6.14.1 | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
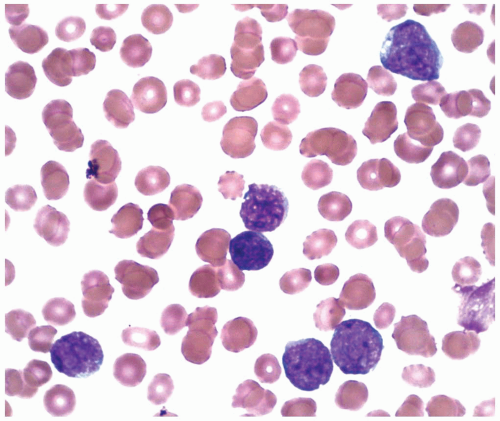 FIGURE 6.14.3 Peripheral blood smear of acute lymphoblastic leukemia (ALL) case shows small, uniform blasts with scant cytoplasm and inconspicuous nucleoli (L1 morphology). Wright-Giemsa, 100× magnification. |
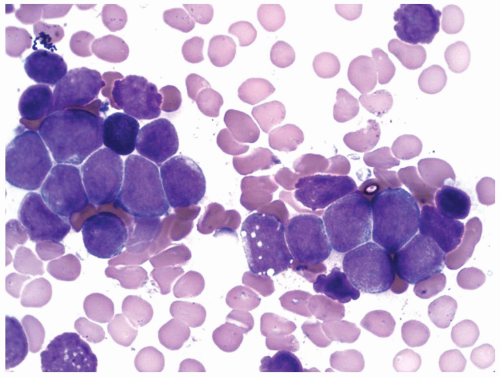 FIGURE 6.14.4 Bone marrow aspirate of acute lymphoblastic leukemia (ALL) case shows large blasts with variable sizes, scant cytoplasm, and inconspicuous nuclei (L2 morphology). Wright-Giemsa, 100× magnification. |
marrow regenerating after chemotherapy or transplantation. It is, therefore, most important to differentiate hematogones from lymphoblasts, particularly in pediatric ALL cases after chemotherapy. Hematogones are small to medium-sized with a high nuclear cytoplasmic ratio, and can mimic small mature lymphocytes or L1 lymphoblasts. Their major morphologic differences from lymphoblasts are the homogeneous nuclear chromatin pattern and the absence of nucleoli (5). In some cases, however, indistinct nucleoli can be present. A low percentage (0.01% to 1.3%) of hematogones has been detected in the peripheral blood of patients without ALL (7).
TABLE 6.14.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 6.14.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 6.14.4 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
constitutes 50% of adult ALL and shows L1 or L2 morphology. It is associated with t(9;22), 11q23 rearrangement, and t(1;19). B-ALL is present in 4% of adult ALL and is the leukemic counterpart of Burkitt lymphoma. Therefore, it shows L3 morphology and is associated with t(8;14), t(2;8), or t(8;22). In B-ALL patients showing no L3 morphology, the cytogenetic changes may also be different, and the prognosis is worse than it is in patients with L3 morphology (9). Because many of these patients carry t(14;18), some authors have suggested that these may represent cases of follicular lymphoma progressing to a leukemic phase with blast transformation (17). However, recent studies show that many of these cases have t(4;11) translocation involving the AF4 and mixed lineage leukemia (MLL) genes (18). The immunophenotype of these cases is characterized by the absence of CD10 and coexpression of myeloid-associated markers, particularly CD15 (19).
tumorigenesis (24,25). The loss of tumor suppressor genes is another mechanism in the pathogenesis of ALL.
TABLE 6.14.5 | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
bone, and lymph nodes are most frequently involved (5). However, any organ system can be affected (Fig. 6.14.5).
TABLE 6.14.6 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
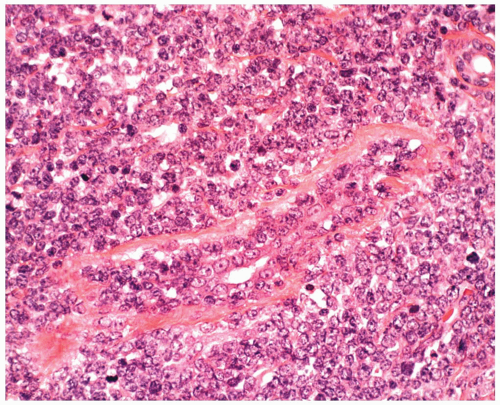 FIGURE 6.14.5 Breast biopsy of acute lymphoblastic leukemia (ALL) case reveals ductal and periductal leukemic infiltration. Wright-Giemsa, 40× magnification. |
In the Working Formulation of non-Hodgkin Lymphoma, LBL was divided into the convoluted and nonconvoluted subtypes (5).
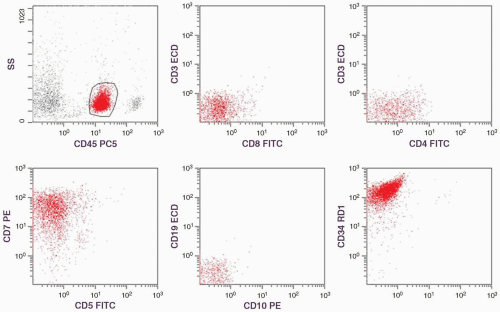 FIGURE 6.15.1 Flow cytometric histograms show that the tumor cells react only to cluster of differentiation (CD)7 and CD34 with low percentages of CD3/CD4 and CD3/CD8. CD5, CD10, and CD19 are negative. SS, side scatter; PC5, phycoerythrin cyanin 5; ECD, phycoerythrin-Texos Red; PE, phycoerythrin; RD1, rhodamine; FITC, fluoresein isothiocyanate. |
are medium-sized with abundant pyroninophilic cytoplasm, which is deeply basophilic and often vacuolated in imprint preparations. Their nuclei are round or ovoid, containing clumped chromatin and multiple nucleoli in tissue sections (10). Touch preparations of a lymph node may show the L3 morphology with more immature-looking chromatin than in tissue sections. In case of doubt, immunophenotyping is helpful; LBL is predominantly of T-cell origin, but Burkitt and Burkitt-like lymphoma are exclusively of B-cell type.
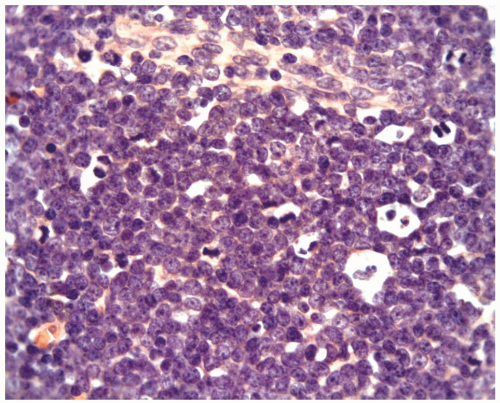 FIGURE 6.15.2 Lymph node biopsy shows a “starry sky” histologic pattern with tangible-body macrophages and mitotic figures in the vacuoles. Hematoxylin and eosin, 60× magnification. |
TABLE 6.15.1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
TABLE 6.15.2 | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
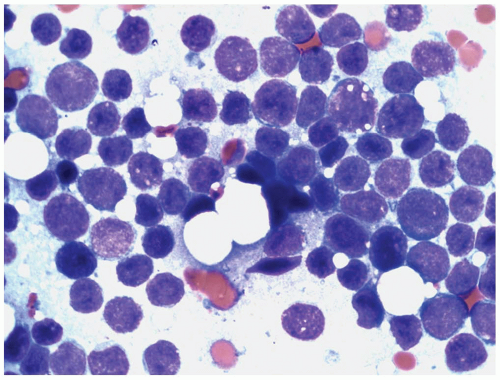 FIGURE 6.15.3 Bone marrow aspirate shows many lymphoblasts with different sizes, immature chromatin pattern, and inconspicuous nucleoli. A few lymphoblasts reveal vacuolated cytoplasm. Wright-Giemsa, 100× magnification. |
TABLE 6.15.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
is only listed in precursor T- and precursor B-LBL and/or leukemia (1). Therefore, a positive TdT reaction may exclude the diagnosis of Burkitt lymphoma. The reactivities of T-cell monoclonal antibodies depend on the stage of thymocytes to which the tumor cells are related (34). Generally, CD2 is consistently positive in all study series, and CD1 is specific for the common thymocyte stage (11,26,27). The reactivities of CD4 and CD8 are usually used as the criteria for stage identification: early thymocytes are CD4−, CD8−; common thymocytes are CD4+, CD8+; and late thymocytes are CD4+, CD8− or CD4−, CD8+.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


