FIGURE 11.1 Evolution of cirrhosis. Fibrosis develops in nonregenerative necrotic areas, producing scars. The pattern of nodularity and scars reflects the type of response to injury (e.g., uniform vs. nonuniform necrosis) and the extent of injury. (From Mulholland MW, Lillemoe KD, Doherty GM, et al. Greenfield’s surgery: scientific principles & practice, 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2010.)
Stellate cells play an important role in the development of cirrhosis. They are found at the interface between the basolateral membranes of hepatocytes and sinusoidal endothelial cells, in the space of Disse. Their normal function is paracrine in nature, regulating hepatocyte and endothelial cells as well as the storage of vitamin A. When hepatocytes are injured, cytokine release results in hypertrophy and proliferation of stellate cells. Eventually, the space of Disse becomes thickened with collagen deposits, and the normal architecture of the fenestrated sinusoidal endothelium is distorted. This leads to vascular distortion and portal hypertension.
Under normal circumstances, portal vein pressure ranges from 5 to 8 mm Hg. Any pressure greater than 8 mm Hg or a hepatic portal venous gradient (HPVG) greater 5 mm Hg is defined as portal hypertension. Practically, this measurement is taken via hepatic vein wedge pressure, similar to a measurement of pulmonary arterial pressure using a Swan-Ganz catheter. The normal venous drainage of the gastrointestinal tract is through the portal vein, into the liver, out of the hepatic veins, into the inferior vena cava (IVC), and then back to the heart. The portal vein is formed by the splenic vein and superior mesenteric vein (SMV). Usually, the inferior mesenteric vein (IMV) joins the splenic vein prior to its junction with the SMV; however, this anatomy varies widely. With obstruction of flow at the presinusoidal, sinusoidal, or postsinusoidal level, portal venous pressure increases and decompression occurs with flow back to the heart via collaterals. These collaterals are generally thin walled and fragile and, with increased flow, become varicosities. Given their fragility, varicosities rupture easily and can cause life-threatening bleeding. Figure 11.2 depicts the collaterals that can form with portal hypertension.
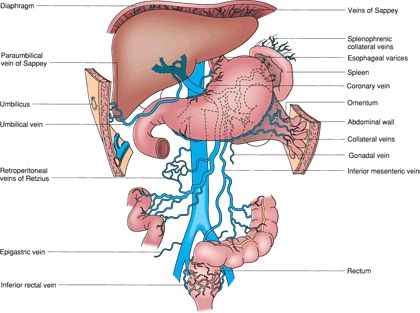
FIGURE 11.2 Potential venous collaterals that develop with portal hypertension. The veins of Sappey drain portal blood through the bare areas of the diaphragm and through paraumbilical vein collaterals to the umbilicus. The veins of Retzius form in the retroperitoneum and shunt portal blood from the bowel and other organs to the vena cava. (From Mulholland MW, Lillemoe KD, Doherty GM, et al. Greenfield’s surgery: scientific principles & practice, 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2010.)
Causes
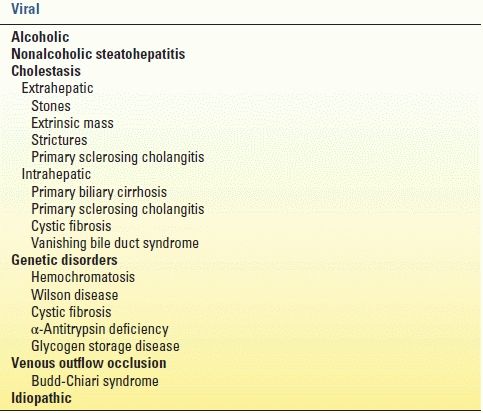
Table 11.1 lists the causes of cirrhosis. The most common cause of cirrhosis in the world is viral hepatitis, but alcohol abuse is the most common cause in the United States. Nonalcoholic fatty liver disease (with progression to nonalcoholic steatohepatitis or NASH) is increasing in prevalence in the United States and is linked with hyperlipidemia, non–insulin-dependent diabetes, and obesity. Immune or inflammatory cirrhosis due to cholestasis is caused by primary biliary cirrhosis and primary sclerosing cholangitis. Hemochromatosis, an inborn error in metabolism, leads to cirrhosis secondary to lipid peroxidation of iron deposits in periportal regions of the liver. Wilson disease is an inherited deficiency in hepatocyte transport of copper into biliary tract, resulting in copper accumulation in the liver, once again causing periportal inflammation and eventual cirrhosis. Obstruction of hepatic venous outflow (Budd-Chiari syndrome) causes sinusoidal congestion and hepatocyte necrosis and, eventually, cirrhosis. Hepatic venous outflow obstruction is associated with “nutmeg” appearance of the liver secondary to multiple small areas of hemorrhage.
TABLE 11.1 Causes of Cirrhosis
DIAGNOSIS
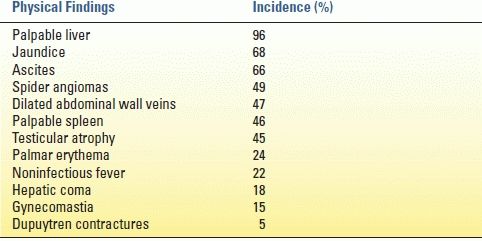
As with any disease, a history and physical examination yield most of the information needed to diagnose a patient with cirrhosis. Clues in the history include acknowledged alcohol abuse, hepatitis, toxin exposure, previous upper gastrointestinal bleeding, hemorrhoids, infections, and increased abdominal girth. Classic physical exam findings in a cirrhotic patient are found in Table 11.2. Fetor hepaticus, bruising, decreased body hair, and purpura are also physical signs associated with cirrhosis.
TABLE 11.2 Physical Findings in Cirrhosis
From Mulholland MW, Lillemoe KD, Doherty GM, et al. Greenfield’s surgery: scientific principles & practice. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2010.
Diagnostic tests are indicated if the history and physical exam raise suspicion for liver disease. Laboratory evaluation includes hepatic panel, complete blood count, chemistry panel, and coagulation profile. As mentioned in earlier sections of this handbook, functional capacity of the liver is evaluated by coagulation studies, platelet count, and bilirubin. Inability of the liver to produce coagulation factors, thrombopoietin, and bilirubin will lead to abnormal values of these labs. Markers of hepatocyte or cholangiocyte damage are AST, ALT, and alkaline phosphatase. The term liver “function” panel traditionally includes AST, ALT, alkaline phosphatase, bilirubin, and lactate dehydrogenase. It is important to understand this misnomer of “function” as these values do not all indicate the liver’s functional capacity. Importantly, abnormal values are not specific for liver dysfunction and may even be normal in the setting of hepatic disease.
In addition to laboratory values, imaging studies are commonly used to further investigate a suspicion of liver dysfunction. Ultrasonography, computed tomography, and magnetic resonance imaging (MRI) can detect cirrhosis. Generally, these studies will reveal a nodular, atrophic liver and splenomegaly. Ultrasound is nearly 90% sensitive and specific for diagnosing cirrhosis. Cirrhotic livers demonstrate multiple nodular irregularities on the anterior surface of the liver distinct from the abdominal wall. Fibrosis can be seen with disruption in parenchyma, though this is often difficult to detect. Computed tomography and MRI are more expensive modalities and do not yield any more information than ultrasound; however, they may reveal an atrophic liver, evidence of venous thrombosis, and/or splenomegaly.
Invasive diagnostic methods (biopsy) definitively establish the diagnosis of cirrhosis; however, in the presence of strong clinical, laboratory, and radiographic evidence, they are not necessary. Direct visualization of the liver during an abdominal surgical procedure can reveal a nodular, atrophic liver. Percutaneous, laparoscopic, or transjugular biopsy, as well as image-guided fine needle aspiration, can be used. Histology will reveal fatty infiltration, balloon-cell degeneration, Mallory bodies, hepatocyte necrosis, fibrosis, or features of cirrhosis.
Two methods of classifying liver disease severity are the Child-Turcotte-Pugh (CTP) score and the Model for End-Stage Liver Disease (MELD) score. These prognostic models of disease severity are important in operative planning and liver transplant allocation. The CTP score is the best predictive score for most patients with cirrhosis and is used to determine risk of morbidity and mortality for surgical procedures (Table 11.3). Originally, the MELD score was developed to determine survival following transjugular intrahepatic portosystemic shunt (TIPS) procedure. It was then adopted as a prognostic indication of a patient’s 90-day survival with optimized medical management. Bilirubin, international normalized ratio, and creatinine are the only values used in calculating the MELD score, so it is a more objective system than CTP score. Because of its objectivity, the MELD score is used as the main determinant of liver transplant allocation in the United States.
TABLE 11.3 Estimated Mortality Rates for Surgical Intervention by CTP Score

CLINICAL MANIFESTATIONS
Renal
Complications of impaired renal function in the setting of cirrhosis are caused by dysregulation of vascular tone. This results in sodium retention, water retention, and ultimately hepatorenal syndrome (HRS) and renal failure. When ascites develops, there is an inability to excrete sodium. Water retention follows sodium retention and is due to the inability of patients with ascites to process free water. Excess water leads to dilutional hyponatremia, which may cause nausea, vomiting, lethargy, and seizures.
HRS is a complex complication of cirrhosis, characterized by renal failure in the absence of intrinsic renal disease. Up to 10% of patients with cirrhosis and ascites develop HRS. Signs of HRS include oliguria, increasing serum creatinine level, rising cardiac output, proteinuria, and decreased arterial pressure. The renin–angiotensin–aldosterone system is overactive, and the renal cortex is markedly vasoconstricted. Prerenal azotemia is difficult to distinguish from HRS, and laboratory values are similar in the two conditions. However, HRS is distinct from acute intrinsic renal failure, with low urine sodium, a high urine/plasma creatinine ratio, high urine osmolality, and normal urine sediment. In an attempt to distinguish HRS from other etiologies, diagnostic criteria for HRS were developed (Table 11.4).
TABLE 11.4 Diagnostic Criteria for Hepatorenal Syndromea
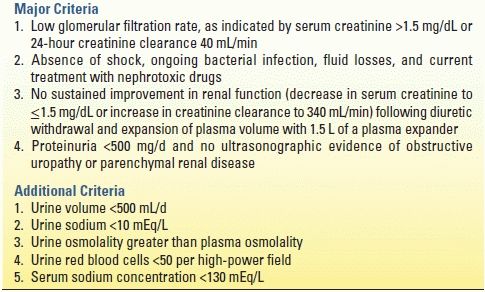
aAll major criteria must be present for the diagnosis of HRS. Additional criteria are not necessary for the diagnosis but provide supportive evidence.
From Arroyo V, Ginés P, Gerbes A, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. Hepatology 1996;23:164, Mulholland MW, Lillemoe KD, Doherty GM, et al. Greenfield’s surgery: scientific principles & practice. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2010.
Pulmonary
Although some causes of cirrhosis can also cause pulmonary complications, cirrhosis itself can have effects on the pulmonary system. As ascites develops, lymphatic transdiaphragmatic communication can result in hepatic hydrothorax. The resultant effusion can directly compress pulmonary parenchyma, which impairs gas exchange and may result in hypoxemia. Hepatopulmonary syndrome (HPS) is diagnosed with hepatic dysfunction, oxygen tension less than 70 mm Hg or diffusion gradient greater than 20 mm Hg, and pulmonary vascular dilation in structurally normal lungs. Patients present with shortness of breath when moving from supine to upright position (platypnea) and dyspnea in the absence of primary pulmonary disease. On physical exam, patients may have clubbing and cyanosis of nail beds. Portopulmonary hypertension is defined by a mean pulmonary arterial pressure of greater than 25 mm Hg in the presence of liver disease. This rare entity has a significant risk of mortality and is a relative contraindication to liver transplant. The exact mechanism of HPS is not known; interestingly, portal hypertension is not required for its development.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


