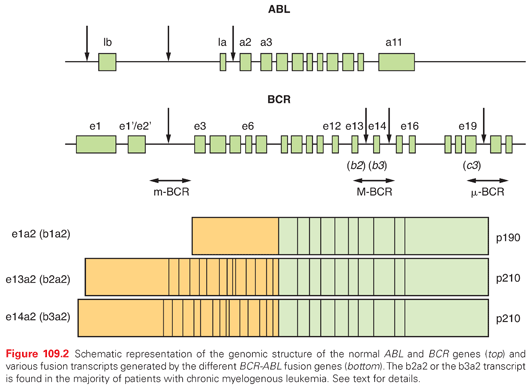
The (9;22) translocation transposes the ABL (Abelson) protooncogene from chromosome 9 into a relatively small, 5.8-kb genomic region on chromosome 22 named the breakpoint cluster region (bcr).11 Although the genomic breakpoints in the ABL gene are highly variable, they almost always occur upstream of the second exon (a2), resulting in translocation of all but exon 1 of ABL. Two slightly different chimeric BCR-ABL genes are present in most patients with CML, depending on the precise location of the breakpoint in the BCR gene. Breaks can occur between exon e13 (also known as b2) and exon e14 (b3), yielding an e13a2 fusion messenger RNA (mRNA), whereas a break occurring between exons 14 and 15 produces an e14a2 fusion mRNA (Fig. 109.2).9 In the majority of patients, either e13a2 or e14a2 transcripts are present, but occasionally, patients have both transcripts in their leukemia cells. Although the e14a2 mRNA encodes a BCR-ABL protein that is 25 amino acids larger than that encoded by the e13a2 transcript, both are referred to as p210BCR-ABL. Patients with e13a2 or e14a2 transcripts have similar prognoses. In 5% of the patients, the breakpoint occurs in other regions of the BCR gene resulting in the so-called rare transcripts; these transcripts have the same biological activity as the more common ones.1,9 It is important, however, to identify patients harboring these rare transcripts because molecular monitoring may otherwise prove impossible.
The BCR-ABL fusion protein resides in the cytoplasm and has constitutive tyrosine kinase activity compared to the tightly regulated activity of the normal ABL product (p145).12,13 The BCR-ABL tyrosine kinase binds to and phosphorylates numerous intracellular proteins. The net effect of this is to induce all of the phenotypic abnormalities observed in patients with CML.9 These include increased proliferation or decreased apoptosis of hematopoietic stem or progenitor cells leading to a massive increase in myeloid cell numbers and the premature release of immature myeloid cells into the circulation, postulated to be due to a defect in adherence of myeloid progenitors to marrow stroma and genetic instability resulting in disease progression. Despite the complexity of BCR-ABL signal transduction, all of the transforming functions of BCR-ABL depend on its tyrosine kinase activity, making this disease an ideal candidate for therapy directed against this activity.
Clinical Manifestations
Of patients with CML, 90% are diagnosed in the chronic or stable phase. Of patients in older studies, 10% to 20% and as many as 50% in more recent series present without symptoms and are diagnosed as a result of finding an elevated white blood count on routine blood sampling. The most common presenting symptoms of CML are related to anemia, splenomegaly, and increased cell turnover. These symptoms include fatigue, left upper quadrant pain, abdominal distention or discomfort, early satiety, weight loss, and night sweats.14
Occasionally, patients may present with a hyperviscosity syndrome, which requires leukapheresis, with manifestations such as stroke, priapism, stupor, or visual changes caused by retinal hemorrhage. The most common physical finding is splenomegaly, its magnitude correlating with the degree of leukocytosis. Ecchymoses are frequently observed, but spontaneous bleeding is uncommon. A lymphadenopathy is not usually seen in the chronic phase.
Laboratory Tests
Peripheral Blood and Bone Marrow
The diagnosis of CML is frequently suspected from an examination of the peripheral blood and bone marrow. The white blood cell (WBC) count in the chronic phase of CML usually exceeds 50 × 109/L at the time of diagnosis and can range up to 800 × 109/L. During the chronic phase, leukemic WBCs differentiate and function normally. The peripheral blood smear shows a full spectrum of myeloid cells from blasts to neutrophils, with blasts comprising less than 15% and usually less than 5% of the WBC differential. Basophilia is invariably present, and its absence should prompt consideration of other myeloproliferative disorders. Eosinophilia is also commonly present. The majority of patients have thrombocytosis and, on occasion, the platelet count may be more than 1000 × 109/L. Most patients with CML have a normochromic, normocytic anemia that is inversely proportional to the degree of leukocytosis.
The bone marrow in patients with chronic phase CML is markedly hypercellular, with a predominance of myeloid cells with full maturation. Blasts are fewer than 15% and most commonly fewer than 5%, and basophilia is also present. Megakaryocytes are usually increased in number, are characteristically small, are hypo- or monolobated, and have a tendency to cluster. Erythroid hypoplasia is frequently present and may seem exaggerated because of the increased myeloid-to-erythroid ratio. Erythroid precursors are otherwise morphologically unremarkable. Reticulin fibrosis is usually absent or mild but may become more prominent with disease progression.15
Cytogenetics
A cytogenetic analysis of 20 bone marrow metaphases has been the standard method to detect the Ph chromosome, which is present in the majority of cells at diagnosis. Although most patients have a typical t(9;22)(q34;q11), approximately 5% have variant translocations that have no impact on prognosis.16 These variant translocations may be simple, involving chromosome 22 and a chromosome other than chromosome 9, or they may be complex, involving one or more other chromosomes in addition to chromosomes 9 and 22.17 Clonal cytogenetic abnormalities in addition to the Ph chromosome are present at diagnosis in approximately 5% of patients diagnosed in chronic phase.18,19 The most common are duplication of the Ph chromosome and trisomy 8, iso-17q, and trisomy 19.18,19 These abnormalities have only a small adverse prognostic impact when identified at diagnosis.19
Molecular Testing
The diagnosis of CML requires the presence of BCR-ABL. In 95% of patients, its presence can be inferred by the detection of the Ph chromosome using standard cytogenetics. Another 5% of patients with a hematologic picture resembling CML who lack a detectable Ph chromosome will have a BCR-ABL fusion gene detectable by fluorescence in situ hybridization (FISH) or reverse transcription–polymerase chain reaction (RT-PCR). These Ph chromosome–negative, BCR-ABL–positive patients have a clinical course that is indistinguishable from that of Ph chromosome–positive, BCR-ABL–positive patients.20 Patients with a hematologic picture resembling CML, but who are Ph chromosome negative and BCR-ABL negative, are classified within the myeloproliferative neoplasm group.21 Some of these patients have mutations in the SETBP122 or CSF3R23 genes, whereas others remain genetically unclassified.
FISH detects the colocalization of large, fluorescently labeled genomic probes specific to the BCR and ABL genes. FISH can be performed on metaphase or interphase cells and on peripheral blood. At diagnosis, when typically 90% of cells are BCR-ABL positive, FISH is a highly accurate diagnostic test, because false-negative results are uncommon.24 However, because of the random colocalization of the signals from the BCR and ABL probes, 8% to 10% of normal cells score positive, making FISH less useful at low disease burdens. A lower false-positive rate can be obtained with dual-FISH (D-FISH), which uses probes that span the breakpoint region.25
RT-PCR to amplify the unique sequences created by the fusion of BCR and ABL is a highly sensitive technique that is ideal for the detection of minimal residual disease.24 PCR testing can either be qualitative, providing information as to the presence or absence of the BCR-ABL transcript, or quantitative, assessing the amount of BCR-ABL message. Quantitative RT-PCR is preferred for monitoring and may allow for the early detection of resistance to therapy.26 False-positive and false-negative results are both possible with RT-PCR, and rigorous controls are required to detect these instances. False-negative results can be due to poor quality RNA, failure of the reaction or failure of the PCR primers to detect rare transcripts, whereas false-positive results are usually due to contamination of the sample.
Differential Diagnosis
The diagnosis of CML is relatively straight forward. The presence of a WBC count over 50 × 109/L with a peripheral blood smear showing a full spectrum of myeloid lineage cells plus basophilia should raise the suspicion of CML. The diagnosis of chronic phase CML can be confirmed by the presence of the BCR-ABL gene as described in the Cytogenetics section and the Molecular Testing section and by the absence of advanced phase features described later in Advanced Phase Disease.
The main differential diagnosis includes a leukemoid reaction and other myeloproliferative neoplasms. In patients with a leukemoid reaction, which is typically seen in patients with underlying infections, the WBC count is usually less than 50 × 109/L and the peripheral blood smear consists predominantly of segmented neutrophils and bands, often with toxic granulations. Less mature myeloid cells are rarely seen; there is no basophilia; and the Ph chromosome and BCR-ABL are absent. Approximately 5% of patients with CML present with extreme thrombocytosis and a minimally elevated WBC count, resembling essential thrombocytosis, but are distinguished by the presence of the BCR-ABL gene. The differential diagnosis with other myeloproliferative neoplasm is relatively easy because the latter frequently lack the basophilia that is seen with CML and lack the BCR-ABL gene, but may have other molecular abnormalities such as SETBP1 or CSF3R mutations.
Historically, the progression of CML to blast crisis occurred in 5% to 10% of patients in the first 2 years after diagnosis and, thereafter, the annual progression rate increased from 20% to 25%. The Sokal score was developed to predict the probability of disease progression, but this and a variety of other factors are now being used to predict the probability of optimal response to therapy. The Sokal score was developed to predict the probability of disease progression.27 Despite it being developed in a pre-TKI era and as a predictor for disease progression, it has been found useful to predict the probability of achieving an optimal response to imatinib.28,29; however, it has been less useful for predicting responses to newer TKIs.30,31 The Sokal score is a numerical value that is calculated from a complex equation that takes into account four factors at diagnosis: the percentage of blasts in peripheral blood, platelet count, spleen size (centimeters below the costal margin) and age of the patient.27 Patients then were divided according to their individual numerical value into three risk groups: low, intermediate, and high, risk of progression, with median survivals of 5, 4, and 3 years, respectively. Many of these patients received therapies not currently in use, such as busulfan and hydroxyurea; however, the Sokal score remains useful to predict the response of patients treated with imatinib, but less so with newer TKIs.28,31 Other variables closely related with the Sokal score such as hemoglobin and white blood count at diagnosis also have prognostic value.29,32,33
Assessment of Response
Response to therapy in patients with CML is assessed by the monitoring of blood counts, cytogenetics, and real-time quantitative polymerase chain reaction (RQ-PCR). A complete hematologic response (CHR) indicates the normalization of the complete blood count (CBC), spleen size, and resolution of symptoms related to the CML. A partial cytogenetic response corresponds to 1% to 35% of Ph-positive metaphases and a complete cytogenetic response (CCyR) is the absence of the Ph chromosome on conventional cytogenetic analysis; a major cytogenetic response (MCyR) is the combination of partial and CCyR.
RQ-PCR has emerged as the most effective and efficient manner to assess leukemic burden because it can easily be done on peripheral blood. At diagnosis, an untreated patient may have in excess of 1012 cells. With a CHR, this falls to approximately 1011 cells and, with a CCyR, to approximately 1010 cells. Because the majority of newly diagnosed CML patients treated with TKIs obtain a CCyR, RQ-PCR is particularly useful for monitoring minimal residual disease because its dynamic range extends two-logs or more below a CCyR. RQ-PCR measure BCR-ABL transcripts in relation to a control gene such as normal ABL or GUSB, and can be expressed as the absolute ratio or as log reductions from the 100% value. In an attempt to introduce some level of uniformity, many laboratories are using the International Scale (IS), whereby values are corrected to those obtained in a reference laboratory.34 It is generally accepted that CCyR corresponds to an approximately 2-log reduction in transcript levels or 1% IS.35 Major molecular response (MR3) is defined as a 3-log reduction in transcript levels or 0.1% IS, and this should correspond to a leukemia burden of approximately 109 cells. Similarly, MR4 is defined as a 4-log reduction (0.01% IS, or 108 leukemic cells). A complete molecular response (CMR) is generally defined as the absence of detectable BCR-ABL transcripts, a status that depends on the technology and criteria for negativity employed, with the limits of technology being somewhere between MR4 and MR5.
Treatment of Chronic Phase Disease
BCR-ABL TKIs are the standard frontline therapy for newly diagnosed patients with chronic phase CML. Hydroxyurea, a well-tolerated oral agent that inhibits DNA synthesis by inhibiting ribonucleotide reductase, remains in use as an initial therapy to control blood counts pending definitive diagnosis and therapy. Allogeneic HCT, although curative, is typically reserved for patients with resistance to TKI therapy or in the management of advanced phase disease due to its morbidity and mortality. Interferon-α, a previous mainstay of therapy, has been supplanted by TKIs, and has been used with variable results in clinical trials attempting to improve molecular responses to TKIs.
BCR-ABL Tyrosine-Kinase Inhibitors
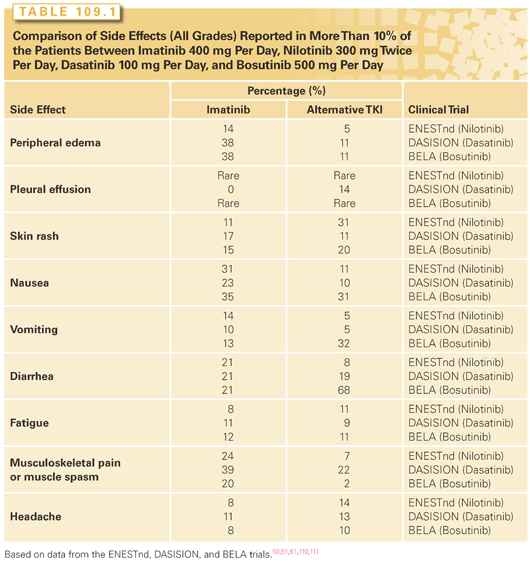
BCR-ABL TKIs inhibit the BCR-ABL tyrosine kinase by competing with ATP binding to the kinase. The first drug of this class was imatinib, which rapidly became the treatment of choice for patients with chronic phase CML after it was approved by the U.S. Food and Drug Administration (FDA) in 2001.2 Today, imatinib, nilotinib, dasatinib, bosutinib and ponatinib are licensed for use in CML. They differ in their respective affinity for the ABL-binding pocket—for example, imatinib has a BCR-ABL IC50 of 221 nM, whereas ponatinib has an IC50 of 0.37 nM and respective half-lives, ranging from 13 hours for imatinib to 4 hours for dasatinib. They also differ in their ability to inhibit other tyrosine kinases, with ponatinib, dasatinib, and bosutinib being the most promiscuous, whereas imatinib and nilotinib are the most specific. Other kinases inhibited by imatinib and nilotinib include the platelet-derived growth factor receptors KIT, and ARG (ABL-related gene), whereas the other drugs inhibit SRC family members amongst many others. These differences explain, at least in part, the different toxicity profiles of the various drugs.36–40 Despite that, it is possible to define a drug class side effect profile that includes myelosuppression, fatigue, gastrointestinal disturbances, hepatotoxicity, myalgias, arthralgias, and skin rashes.41 All five drugs can cause these side effects, but the relative frequency of the individual side effect differs for each drug. Common toxicities observed are listed in Table 109.1.
Imatinib
With over 15 years of experience and hundreds of thousands of patients using imatinib as first-line therapy, the side effects are well understood and there are no apparent long-term safety concerns. Approximately 70% of patients that receive imatinib as first-line therapy will achieve CCyR by 12 months, and 80% will have done so by 5 years.28,29 The 8-year survival for 553 patients treated with imatinib as part of a randomized trial comparing imatinib to interferon plus Ara-C was 85%, or 93% if only CML-related deaths were considered. This high survival rate has been confirmed in additional single-center studies.42 Imatinib is given once daily, normally with food (in order to prevent nausea). The current standard dose of imatinib is 400 mg per day. Several randomized studies comparing 400 mg per day to 800 mg per day in newly diagnosed patients revealed more rapid responses with the higher doses; however, one-third of patients required dose reduction due to greater toxicity. With longer follow-up, the response rates to 400 mg per day and higher doses are similar. Progression-free survival (PFS) was not impacted by the higher doses, although a recent study from Germany has suggested that deeper molecular remissions that are more frequently obtained with higher doses of imatinib may translate into a trend toward improved PFS.43–45
Nilotinib
Nilotinib is a BCR-ABL inhibitor that was rationally designed to be more potent and selective than imatinib. Until recently, it had been used as a second-line agent at a dose of 400 mg twice daily. At this dose, it induces CCyR in 30% to 40% of the patients who are resistant to imatinib, and most interestingly, there is no cross-intolerance with imatinib.46–48 Nilotinib obtained a license for first-line use in 2010. The first-line dose is 300 mg twice daily. Nilotinib is administered twice daily in a fasting state because food increases absorption and may lead to increased side effects, particularly the prolongation of Qtc intervals. Overall, nilotinib is well tolerated and causes less nausea, myalgia, arthralgia, and fluid retention than imatinib. On the other hand, it produces skin rashes or pruritus in the majority of patients; in most cases, these can be easily controlled with antihistamines. Nilotinib induces hyperglycemia in approximately 40% of patients; it also causes increases in cholesterol and triglyceride levels. Hepatotoxicity is frequent, although this is normally limited to mild increases in transaminases that do not require action. A severe toxic hepatitis occurs rarely. Similarly, nilotinib causes an increase in the bilirubin level in the majority of patients, but this seldom necessitates any modification of therapy. Nilotinib has also been associated with progressive peripheral arterial occlusive disease,49 although the incidence of this complication is not yet clear.
Dasatinib
Dasatinib is a multitarget kinase inhibitor that is more than 300 times more potent than imatinib in inhibiting the BCR-ABL oncoprotein in vitro.50,51 As with nilotinib, most of the experience to date has been using dasatinib in patients who are resistant to imatinib. Initially, dasatinib was used at a dose of 70 mg twice daily due to its short half-life, but a large randomized study comparing several schedules of dasatinib in patients with imatinib resistance showed that 100 mg once daily was equally efficacious to the 70 mg twice daily or 140 mg once daily schedules, but with significantly less toxicity for chronic phase patients.52 Dasatinib induces CCyR in 30% to 40% of patients with imatinib resistance.48,53,54 Dasatinib was also approved for first-line use in chronic phase CML patients at a dose of 100 mg per day. Dasatinib is generally well tolerated. Pleural effusions are the main complication of dasatinib therapy. Most studies report an incidence below 25%, although rates as high as 54% have been reported.51,55–58 Pleural effusions can occur at any time during therapy and often recur despite a dose reduction. Diagnostic thoracocentesis is not usually required and, in practice, the effusion nearly always resolves on discontinuing the drug. Resolution of the pleural effusion can be accelerated by using 0.5 mg per kilogram of prednisolone for 1 or 2 weeks. Dasatinib has been associated with pulmonary hypertension. The incidence of this complication is not clearly established, and it may be at least partially reversible on discontinuation of the drug.59
Bosutinib
Bosutinib is a dual SRC and ABL TKI, which is currently licensed only for second-line use. Similarly to nilotinib and dasatinib, bosutinib induces CCyR in 30% to 40% of the patients who are resistant to at least one prior TKI therapy.60 Bosutinib has also been evaluated in newly diagnosed patients, but it did not meet its primary endpoint of improved rates of CCyR at 1 year.61 The drug is well tolerated. Its main side effect is diarrhea, which can easily be managed symptomatically.60–62 Bosutinib has also been found to cause pleural effusions and pulmonary hypertension, although the incidence of both complications appears to be lower than with dasatinib.
Ponatinib
Ponatinib is the latest addition to the TKI armamentarium. It has been licensed for the management of patients who are resistant to at least two prior TKIs or who harbor the kinase domain mutation T315I. This mutation confers resistance to all the other TKIs discussed in this chapter. The peculiar chemical structure of ponatinib allows it to overcome the steric hindrance caused by the substitution of a threonine by isoleucine in position 315 of the kinase domain.63 The efficacy of ponatinib has been explored in the PACE study where approximately 200 chronic phase patients who were resistant or intolerant to either dasatinib or nilotinib (the majority of these patients had failed at least three prior TKI lines) and 64 chronic phase patients with the T315I mutation were treated with 45 mg daily. The CCyR rate was 46% with a higher response rate in patients with the T315I mutation.64–66 The main side effects are skin rash, pancreatitis, and hepatotoxicity. Various cardiac, cerebral, and peripheral vascular thrombotic events have been reported in patients on ponatinib. These have occurred in up to 27% of patients treated with ponatinib and have included fatal myocardial infarction and stroke. These vascular side effects led to the early termination of the phase 3 trial comparing ponatinib with imatinib as frontline therapy and a recommendation that lower doses of ponatinib be used—30 mg in patients initially and 15 mg after patients achieve a MR3. To date, strategies combining ponatinib with antithrombotic agents have not been formally reported.
Choice of Initial Therapy for the Newly Diagnosed Patient in Chronic Phase
Imatinib, nilotinib, and dasatinib are licensed for first-line use. Four randomized studies have been conducted comparing imatinib to newer TKIs, one each comparing imatinib to nilotinib or bosutinb, and two comparing imatinib to dasatinib. Data from these studies are summarized in Table 109.2. A few generalities emerge from these studies. All therapies are extremely effective with excellent PFS. The newer, more potent TKIs induce faster cytogenetic and molecular responses, but in only the imatinib versus nilotinib study has this translated into a significant difference in PFS at 3 years. In the other studies, there was either no difference or a trend to improved PFS, but no studies have shown a significant difference in overall survival (OS). With most relapses on imatinib occuring in the first 3 years, it is unlikely that additional follow-up will lead to significant changes in this data.
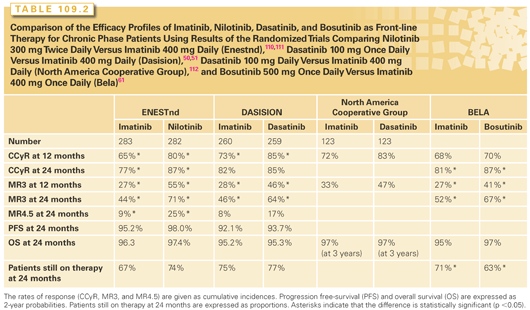
Imatinib is still the first-line TKI for many clinicians because its long-term side effect profile is well understood. Further, in most health systems, it is significantly cheaper than dasatinib or nilotinib. The main arguments in favor of using nilotinib or dasatinib as first-line therapies are that they induce deep molecular responses in a higher proportion of patients and that this may impact PFS. With optimal responses to imatinib being highly dependent on the Sokal score, it would be reasonable to choose nilotinib or dasatinib for patients with higher risk Sokal scores.
Another consideration for the selection of initial therapy is comorbid conditions based on the side effect profiles described in Table 109.1
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


