Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is a neoplastic disease characterized by the accumulation of monoclonal lymphocytes in blood, bone marrow, and lymphoid tissues. These lymphocytes are small, mature-appearing B cells typically expressing CD19, CD5, and CD23. It is generally a disease of older people and prognosis ranges widely from a few years to many years, but it is not considered curable outside of the bone marrow transplant setting. At times these neoplastic cells predominate in lymph nodes leading to the classification as a lymphoma. Hence, the WHO in 2008 has defined this neoplasm as chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) (1).
EPIDEMIOLOGY
CLL is the most common form of leukemia among adults of Western societies and accounts for 30% of all leukemias. In the United States, 16,060 new cases or 4.2 per 100,000 persons and 4580 deaths are projected for 2012 (2, 3). The male-to-female ratio is approximately 3:2. CLL accounts for 1% of all cancers, and is a disease of older adults with a median age of 72 years; only 10% of patients are <50 year old. The disease tends to run in families. When multiple members of a family have CLL, a detectable clone of CLL cells can be found by flow cytometry in 13.5% of apparently healthy first-degree relatives of patients. Also, among normal individuals >40 years of age, a clone of B cells consistent with CLL can be found by multiparameter flow cytometry in 3.5% of subjects (4). It is uncertain whether these individuals will progress to clinically significant disease. CLL is uncommon in Asia.
BIOLOGY
 CAUSE
CAUSE
The cause is unknown. Environmental factors such as exposure to radiation, sunlight, chemical toxins, or viruses are not associated with an increased incidence of the disease. HLA haplotype is not associated with disease susceptibility.
 MOLECULAR DEFECT
MOLECULAR DEFECT
CLL cells are characterized by a defective B-cell receptor (CD79a and CD79b) that does not respond properly to antigen engagement but is associated with constitutive signaling intracellularly through immuno-receptor tyrosine-based activation motifs (ITAMs) to activate a cascade of kinases including Lyn and Syk leading to proliferation, inhibition of apoptosis, or, on occasion, promotion of apoptosis (5, 6). These changes lead to an accumulation of CLL cells in G0. CLL cells derive from antigen-experienced B lymphocytes and have the phenotype of activated cells.
During an immune response, normal B cells encountering antigen will travel to a germinal center and undergo a series of point mutations in the immunoglobulin genes, which result in a more snug fit for the antigen in its binding site. These somatic mutations can be detected by sequencing the immunoglobulin heavy-chain variable-region (IgVH) genes. Patients with CLL cells that contain somatic mutations in their Ig genes (a little over 50%) will have a much better prognosis than patients with CLL cells containing germline Ig sequences. Two surrogate markers for mutational status are more easily obtainable than mutational analysis: CD38, a cell-surface enzyme involved in regulating B-cell activation, and ZAP-70, the 70-kD zeta-associated protein normally found in T cells and NK cells (5). CD38 levels tend to be high in CLL cells bearing unmutated Ig genes and can be easily assayed by routine flow cytometry. The intracellular protein ZAP-70 can also be assayed by flow cytometry, but it is technically more difficult. Elevated levels of ZAP-70 are also associated with CLL expressing unmutated Ig genes.
CLL cells can develop other cytogenetic abnormalities. Commonly detected clonal evolutions involve DNA deletions at chromosomes 13q14, 11q22-23, 17p13, and 6q21.
 IMMUNE DYSREGULATION
IMMUNE DYSREGULATION
Patients with CLL frequently demonstrate immune dysregulation ranging from hyperreactivity to external stimuli such as insect bites to frank immunodeficiency with frequent infections. CLL cells elaborate immune suppressive cytokines such as CD27 or transforming growth factor-γ, which impede immune activation. CLL cells can also downmodulate CD40 ligand on CD4 T cells, which results in defective function of T cells as well as the initial steps in immunoglobulin production. Paradoxically autoimmunity may develop leading to autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura (ITP), pure red cell aplasia, or autoimmune neutropenia. Usually the pathogenic autoantibodies are not produced by the CLL cells but are produced by normal lymphocytes and plasma cells in response to factors produced by the CLL cells.
CLINICAL PRESENTATION
Most patients with CLL present with mild symptoms of fatigue or malaise. Over 25% will present asymptomatically with incidental lymphocytosis in blood and nearly 80% of patients have nontender lymphadenopathy. Occasionally, patients will present with advanced disease with fever, sweats, weight loss, anemia, thrombocytopenia, or recurrent infections. Although computerized axial tomography (CT) scans are not part of the routine evaluation of CLL, they can on occasion demonstrate massive lymphadenopathy not otherwise appreciated. Rarely, lymphadenopathy in CLL will be responsible for organ dysfunction such as ureteral obstruction with hydronephrosis or biliary obstruction. Splenomegaly is present initially in approximately 50% of patients and may cause discomfort or early satiety. Patients may have anemia or thrombocytopenia. It is of critical importance to assess whether the cause is bone marrow infiltration or destruction by autoantibodies, as the treatment approach may vary according to the mechanism. The examination of the peripheral blood smear will generally show a lymphocytosis of mature-appearing lymphocytes with occasional larger lymphocytes and smudge cells. The smudge cells, which are broken lymphocytes resulting from the technique to prepare the blood smear, are characteristic of CLL. Despite very high lymphocyte counts in some patients with CLL, hyper-leukocytosis with pulmonary or cerebral symptoms requiring emergency intervention is rare; patients have generally tolerated lymphocyte counts as high as 800,000/ml without problems of hyperviscosity. Bone marrow biopsies performed at the time of presentation will show infiltrating CLL cells. The pattern of infiltration is said to be prognostic, and four patterns are recognized: interstitial, nodular, mixed, and diffuse (prognosis going from better to worse). A Coombs test is recommended at the time of presentation because approximately 20% of patients will test positive; however, only 8% will have autoimmune hemolytic anemia. Approximately 15% of patients will present with normochromic normocytic anemia that is not associated with anti-RBC antibodies.
DIAGNOSIS
 SPECIFIC CRITERIA
SPECIFIC CRITERIA
The diagnostic criteria for CLL have evolved over time, and currently most clinicians follow the criteria outlined in 2008 by the World Health Organization(WHO)(1) and by the International Workshop on Chronic Lymphocytic Leukemia (IWCLL) (7). Accordingly, the diagnosis of CLL requires a lymphocytosis of >5000/µl with cells that are B cells (positive for CD19, CD20, and CD23) and carry the aberrantly expressed T-cell marker CD5. Monoclonality needs to be demonstrated by surface immunoglobulin restriction to either kappa or lambda light chain. The lymphocytosis must also consist of <55% prolymphocytes, otherwise a diagnosis of prolymphocytic leukemia is favored. In the event that fewer than 5000/ul B-lymphocytes are detected in the peripheral blood, a diagnosis of SLL may be made with the presence of lymphadenopathy and/or splenomegaly. With neither lymphadenopathy nor splenomegaly a diagnosis of monoclonal B-lymphocytosis (MBL) may be made with <5000/ul monoclonal B-cells in the peripheral blood. Although approximately 80% of cases will have an abnormal karyotype by fluorescence in-situ hybridization (FISH) analysis, no specific cytogenetic abnormality defines the disease.
 DIFFERENTIAL DIAGNOSIS (SEE TABLE 29-1)
DIFFERENTIAL DIAGNOSIS (SEE TABLE 29-1)
Benign Lymphocytosis Patients may present with a high lymphocyte count when infected with various organisms such as: viruses, Bordetella pertussis, or Toxoplasma gondii. Usually, the lymphocytosis is not sustained, however, flow cytometry will not show monoclonal surface Ig or expression of CD5 on B cells.
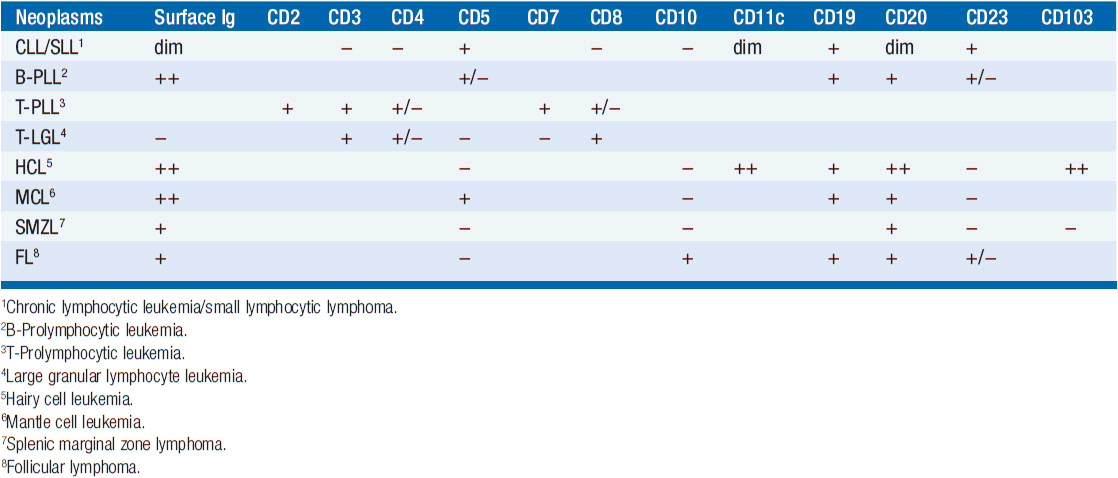
Table 29-1B IMMUNOPHENOTYPE OF CLL AND SIMILAR NEOPLASMS
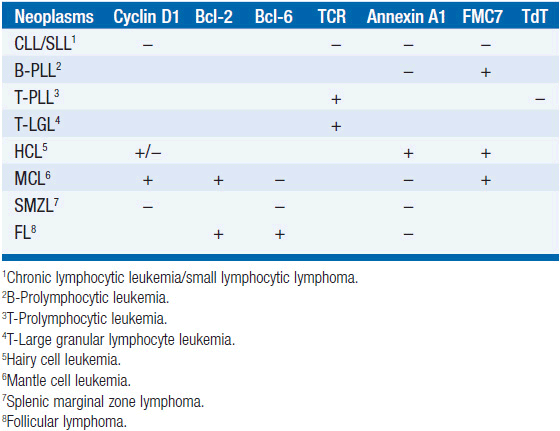
Distinguishing Cll from other B-cell leukemias (WHO criteria) CLL is generally easily distinguished from other monoclonal B-cell malignancies that can be associated with lymphocytosis. CLL is surface Ig weak (others are strong), CD5 positive (others are negative), CD23 positive (others are negative), and CD79b/CD22 weak (others are strong). In addition, the antibody FMC7 does not react with CLL cells but does react with other B-cell leukemias.
B-prolymphocytic leukemia When the percentage of prolymphocytes in blood exceeds 55% in a patient who otherwise makes criteria for CLL, a diagnosis of prolymphocytic leukemia is made. It accounts for 1% of lymphoid leukemias. The prolymphocytes are larger cells being 10–15 μm in diameter (vs 7–10 μm for CLL cells), and they have more cytoplasm, frequently have nucleoli, and stain brightly for surface immunoglobulin. Prognosis is worse for prolymphocytic leukemia.
T-prolymphocytic leukemia (T-PLL) In about 2% of cases of small lymphocytic leukemia, the blood contains small- to medium-sized T-lymphoid cells with nongranular basophilic cytoplasm and round or oval nuclei with a nucleolus. Patients often have hepatosplenomegaly and lymphadenopathy and 20% have skin infiltration. The tumor cells are CD2, CD3, and CD7 positive while TdT and CD1a are negative. In 60%, the cells express CD4 but not CD8; in 15% they express CD8 but not CD4; and in 25% they express both CD4 and CD8. The course of disease is rapid with median survival of 1 year.
T-cell large granular lymphocytic leukemia In 2%–3% of cases of small lymphocytic leukemia, the cells in the peripheral blood have abundant cytoplasm that contains azurophilic granules. These patients usually present with neutropenia and minimal lymphocytosis. Flow cytometry shows these cells to be T cells, being positive for CD3, CD8, and the T-cell receptor (TCR). The cells are positive for CD57 in over 80% of cases and usually negative for CD4. The course of disease is often indolent.
Hairy cell leukemia In about 2% of cases of small lymphocytic leukemia, the blood smear will show a minimal lymphocytosis with lymphocytes demonstrating villous projections. Flow cytometry is diagnostic with cells positive for CD19, CD20, CD22, CD25, CD103, CD11c, and annexin A1. Flow cytometry will be negative for CD5 and CD10. Note that some cells may be positive for cyclin D1. Soluble CD25 levels are elevated. Patients present with splenomegaly and pancytopenia; monocytopenia is often noted. Long remissions have been obtained using cladribine or other nucleosides.
Mantle cell lymphoma with blood involvement These cells in blood and lymph nodes may look very much like CLL cells, and flow cytometry is required for a secure diagnosis. The cells are positive for CD5, FMC-7, and cyclin D1, and they have bright surface immunoglobulin, while being negative for CD10, CD23, and BCL6. Aberrant phenotypes exist, so genetic confirmation is desirable by testing for t(11;14)(q13;q32), which is the translocation of IGH with CCND1, the gene for cyclin D1. Prognosis for mantle cell lymphoma is generally much worse than that for CLL.
Splenic marginal zone lymphoma Patients generally present with splenomegaly and lymphocytosis with the lymphoid cells containing villous projections. Flow cytometry distinguishes these cells, since they are positive for CD19, CD20, CD79a, and surface immunoglobulin. Importantly, the cells are negative for CD5, CD10, CD23, annexin A1, and cyclin D1. The disease usually follows an indolent course.
Follicular lymphoma with blood involvement Examination of the blood smear usually shows these cells to have cleaved nuclei, while CLL cells do not. The flow cytometry pattern shows cells positive for CD10, CD19, CD20, CD22, and surface immunoglobulin with a bright pattern. They are positive for BCL2 and BCL6, while being negative for CD5.
EVALUATION OF THE PATIENT
The following evaluation is commonly performed on patients with CLL:
History and physical examination (B symptoms, lymphadenopathy, splenomegaly)
Complete blood count with differential, reticulocyte count, Coombs test, and LDH
Routine chemistries, uric acid, liver function tests including albumin and globulin
Hepatitis B serologies should be done, if treatment with rituximab is likely
Serum protein electrophoresis
Beta-2-microglobulin
Flow cytometry of blood: CD5, CD10, CD19, CD20, CD23, CD38, cyclin D1
Clinical trials may request ZAP-70 and IgVH mutational analysis Lymph node biopsy: Immunohistochemical panel: CD3, CD5, CD10, CD20, CCD1
Bone marrow biopsy with cytogenetics and FISH analysis: del(11q), trisomy 12, del(13q), del(17p). Consider FISH analysis for t(11;14) when mantle cell lymphoma is suspected
Note that physical examination and peripheral blood examination are usually adequate for initial evaluation of asymptomatic patients, and bone marrow biopsy is usually delayed until treatment is imminent or when indicated to assess the pathophysiologic basis of anemia or thrombocytopenia. Clinical trials may require CT scans to assess response. Note that PET scans are generally not indicated unless Richter’s transformation (histologic progression to diffuse large B-cell lymphoma with an accelerated pace of disease) is suspected.
PROGNOSTIC FACTORS
 STAGING
STAGING
Using clinical data from the physical examination and the complete blood count with differential, an estimate of prognosis can be obtained. The critical features used to assign clinical stage of disease are the presence of lymphadenopathy, splenomegaly, or hepatomegaly, and the presence of anemia and/or thrombocytopenia. The Rai (8) and Binet (9) staging classifications for CLL are shown in Table 29-2. Prognosis varies from 2 years with advanced stage disease to over 10 years with early stage disease.

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


