INTRODUCTION
SUMMARY
Chronic lymphocytic leukemia is a malignancy of mature B cells characterized by progressive lymphocytosis, lymphadenopathy, splenomegaly, and cytopenias. The progressive accumulation of leukemic B cells is a consequence of defective apoptosis and survival signals derived from the microenvironment. Progressive disease results in dysregulation of the cellular and humoral components of the effector immune system with a resultant increase in the incidence of infectious complications, which constitutes the leading cause of morbidity and mortality in this disease. Significant therapeutic advances have been realized in recent years, especially with the development of well-tolerated targeted antibodies and kinase inhibitors. Although not curative, these therapies have resulted in significant improvements in patient outcomes with substantial increases in progression-free and overall survival intervals. Multiple novel agents are also in development with the potential to alter the treatment paradigms for this disease and ultimately to affect a cure.
Acronyms and Abbreviations
ABC, activated B cell; ABVD, Adriamycin, bleomycin, vinblastine, and dacarbazine; ADCC, antibody-dependent cell-mediated cytotoxicity; ADP, adenosine diphosphate; AIHA, autoimmune hemolytic anemia; ALL, acute lymphoblastic lymphoma; ARLTS1, ADP-ribosylation factor-like tumor-suppressor gene 1; ATM, ataxia-telangiectasia mutated; BAK, Bcl-2 homologous antagonist/killer; BCL-2, B-cell lymphoma-2; BCR, B-cell receptor; BiTE, Bi-specific T-cell engaging; BR, bendamustine and rituximab; BTK, Bruton tyrosine kinase; CALGB, Cancer and Leukemia Group B; CAP, cyclophosphamide, doxorubicin, and prednisone; CAR-T, chimeric antigen receptor T cell; CD, cluster of differentiation; CDC, complement-dependent cytotoxicity; CDK, cyclin-dependent kinase; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CIRS, cumulative illness rating scale; CLL, chronic lymphocytic leukemia; CMP, cyclophosphamide, melphalan, and prednisone; CMV, cytomegalovirus; CR, complete response; CRi, complete response with incomplete count recovery; CT, computed tomography; CVP, cyclophosphamide, vincristine, and prednisone; CXCR4, C-X-C chemokine receptor type 4; DAPK, death-associated protein kinase; ERK1, extracellular signal-regulated kinase 1; FC, fludarabine and cyclophosphamide; FCR, fludarabine, cyclophosphamide, and rituximab; FDG-PET, fluorodeoxyglucose positron emission tomography; FISH, fluorescent in situ hybridization; FR, fludarabine and rituximab; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GVL, graft-versus-leukemia; HCL, hairy cell leukemia; HLA, human leukocyte antigen; Ig, immunoglobulin; IGH, immunoglobulin heavy chain; IGHV, immunoglobulin heavy-chain variable region; IL, interleukin; ITK, IL-2–inducible T-cell kinase; ITP, immune thrombocytopenia; IVIG, intravenous immunoglobulins; IWCLL, International Workshop on Chronic Lymphocytic Leukemia; KLHL6, Kelch-like protein-6; LDH, lactate dehydrogenase; LYN, Lck/Yes novel; MBL, monoclonal B-cell lymphocytosis; MCL-1, myeloid cell leukemia-1; MHC, major histocompatibility complex; miRNA, microRNA; MMP, matrix metalloproteinase; MMR, measles, mumps, and rubella; MRD, minimal residual disease; MYD88, myeloid differentiation primary response gene 88; NAD, nicotinic acid adenine; NCCN, National Comprehensive Cancer Network; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor kappa B; NK, natural killer; NOTCH1, Notch homologue 1, translocation-associated; NRM, nonrelapse mortality; OFAR, oxaliplatin, fludarabine, and rituximab; ORR, overall response rate; OS, overall survival; PCR, pentostatin, cyclophosphamide, and rituximab; PCV-13, pneumococcal 13-valent conjugate vaccine; PFS, progression-free survival; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PLCγ2, phospholipase C-gamma-2; PLL, prolymphocytic leukemia; PR, partial response; PR+L, partial response with lymphocytosis; PRCA, pure red cell aplasia; RB, retinoblastoma; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-EPOCH, rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin; R-hyperCVXD, fractionated cyclophosphamide, vincristine, liposomal daunorubicin, and dexamethasone; RS, Richter syndrome; SCT, stem cell transplantation; SDF-1, stromal cell–derived factor-1; SF3B1, splicing factor 3B subunit 1; SLL, small lymphocytic B-cell lymphoma; SNP, single nucleotide polymorphism; STAT3, signal transducer and activator of transcription 3; SUV, standardized uptake value; SYK, spleen tyrosine kinase; TCL1, T-cell leukemia/lymphoma protein 1A; TGF-β, transforming growth factor-β; T-LGL, T-cell large granular lymphoma; TNF-α, tumor necrosis factor-α; TP53, tumor protein p53; T-PLL, T-cell prolymphocytic lymphoma; TRAP, tartrate-resistant acid phosphatase; TRM, transplant-related mortality; VCAM, vascular cell adhesion molecule; XIAP, X-linked inhibitor of apoptosis protein; XPO1, gene encoding exportin-1; ZAP-70, zeta-chain–associated protein kinase of 70 kDa.
DEFINITION AND EPIDEMIOLOGY
Chronic lymphocytic leukemia (CLL) is one of the most common leukemias in the Western hemisphere. CLL is a malignant lymphoid neoplasm that is characterized by the accumulation of a population of small mature B cells. The diagnosis of CLL requires the presence of at least 5000 circulating B cells/μL with clonality demonstrated by flow cytometry according to International Workshop on Chronic Lymphocytic Leukemia (IWCLL) criteria.1 Over the last 2 centuries, significant strides have been made in the understanding of the disease pathophysiology, clinical features, and complications arising from CLL. CLL was initially described by Virchow in the 1840s when he described patients with lymph node enlargement and leukocytosis. Subsequent studies revealed the involvement of the spleen and marrow and led to the introduction of the term “lymphosarcoma.” Ensuing natural history studies established the malignant and clonal nature of the disease and categorized patients based on clinical presentation. Surveillance, Epidemiology, and End Results Program (SEER) data from 2013 estimate the prevalence of CLL in the United States at 126,553 patients, of whom 72,569 are males. The American Cancer Society estimates 15,720 new cases of CLL in 2014 with a median age of diagnosis of 72 years. This cancer is more common in men,2 uncommon in patients younger than the age of 40 years, and extremely rare in children. The risk also increases progressively with age3 and decreases with increasing parity in women.4 It is also relatively uncommon in Asians,5 even in Asian immigrants to the Western hemisphere,6 suggesting a possibility of a genetic predisposition. In the last few years there has been a tremendous growth in the understanding of the disease biology, which has resulted in the development of numerous new therapeutic options with resultant transformation in the management of this illness. Despite the significant improvement in the prognosis of this disease, cure currently remains elusive.
ENVIRONMENTAL FACTORS
Multiple studies have been conducted in an attempt to identify environmental factors that predispose people to the development of CLL. These studies have consistently identified a family history of hematologic malignancies as a strong predictive factor for the development of CLL.7,8 In the reported International Lymphoma Epidemiology Consortium (InterLymph) Non-Hodgkin Lymphoma Subtypes Project,9 detailed correlative studies were performed on a large cohort of white patients with CLL as compared to normal controls. The InterLymph study identified multiple factors that were associated with the presence of CLL, including: (1) family history of a first-degree relative with hematologic malignancy including lymphomas, leukemias, and myeloma; (2) a history of working or living on a farm; (3) hairdressers; and (4) a history of hepatitis C infection. Factors that were found to be protective include a history of allergies, blood transfusions, sun exposure, and smoking. CLL is also recognized as a service-connected illness among Vietnam War veterans who were exposed to Agent Orange.10 Limited data suggests a possible risk of CLL in individuals chronically exposed to electromagnetic fields.11,12 Radiation exposure, however, has not been shown to correlate with the development of CLL as revealed by population-based studies on survivors of the Hiroshima atomic bomb and studies on nuclear reactor workers.13,14 A smaller study conducted on survivors of the Chernobyl nuclear power plant accident did, however, suggest a slightly higher incidence of CLL in these people.15
HEREDITARY FACTORS
CLL has a strong familial predisposition with up to 10 percent of patients with a first- or second-degree relative with CLL and an even higher percentage when also considering individuals with monoclonal B-cell lymphocytosis.16,17 Risk of acquiring CLL is also potentially increased in patients with first-degree relatives with other indolent non-Hodgkin lymphomas including lymphoplasmacytic lymphomas.18 Death-associated protein kinase (DAPK) and CD57 (LEU7) germline mutations have been linked to familial predisposition in a single CLL family.19 Association studies have identified multiple putative genes, polymorphisms, and genetic factors including CD5,20 CD38,21 tumor necrosis factor (TNF)-α,22 and human leukocyte antigen (HLA) haplotypes,23 among others,24 but definite mechanistic studies demonstrating clear contribution to pathogenesis are lacking.
DISEASE BIOLOGY
CLL has varied presentations and complex biology that is the focus of ongoing studies of particular relevance to the practicing oncologist. CLL cells are derived from the B-lymphocyte lineage as demonstrated by their expression of the pan–B-cell surface markers including CD19, and a weaker expression of CD20.25,26 Furthermore, CLL B cells express the memory B-cell marker CD27,27 and also exhibit similar microarray profiles, suggesting a potential relationship to the normal memory B cell.28,29 Most CLL B cells also express κ and λ immunoglobulin light chains on their surface, along with M and D immunoglobulin heavy chains.30,31 These immunoglobulins are often reactive toward self-antigens and polyreactive,32,33 and may play a role in the survival and expansion of the leukemia cell clone.
CLL is characterized by gradual accumulation of leukemic cells primarily from defective apoptosis that is partly contributed by microenvironment interaction. Overexpression of multiple antiapoptotic proteins like BCL-2 (B-cell lymphoma-2), MCL-1 (myeloid cell leukemia-1),34,35 BAK (Bcl-2 homologous antagonist/killer), and XIAP (X-linked inhibitor of apoptosis protein) along with transcription factors like NF-κB (nuclear factor kappa B), NFAT (nuclear factor of activated T cells), and STAT3 (signal transducer and activator of transcription 3) have been clearly demonstrated in CLL.36 Additional survival signals are provided by the microenvironment and include cellular factors like nurse-like cells,37 and various chemokines like CXCR4 (C-X-C chemokine receptor type 4) and SDF-1 (stromal cell–derived factor-1).38 A combination of these factors results in providing the CLL cells with a survival and proliferative advantage. CLL B cells exhibit differential proliferation in the various disease compartments, including the blood, spleen, and marrow.39,40 CLL B cells isolated from the blood of patients lack proliferative potential in vitro and are restricted to the resting phase of the cell cycle.41,42 These cells also undergo spontaneous apoptosis in routine culture conditions. Their survival can be extended when these cells are cultured on stromal cells or nurse like cells that are generally found in the secondary lymphoid organs.43,44 These secondary lymphoid organs are generally diffusely infiltrated by the B cells and are potentially the sites of cell division and proliferation.45 In vivo, the leukemic cell clones increase by 0.1 to 1 percent per day despite a stable blood lymphocyte count as assessed by elegant heavy water studies.46
IMMUNE DYSREGULATION
CLL is characterized by progressive immune dysregulation both in the cellular and humoral compartments.47 Progression of CLL is associated with an early increase in the absolute number of circulating T cells and specifically an increase in the immunosuppressive T-regulatory cells.48,49 Functional studies on T cells from patients with CLL have also shown the T cells to be anergic and with impaired proliferative potential, but with a retained capacity to produce cytokines.50 Functional defects have also been observed in granulocytes.51 The leukemic B cells are responsible for initiating and propagating the immune dysregulation observed in the disease by producing immunosuppressive cytokines like transforming growth factor-β (TGF-β) or by downregulating critical surface molecules required for development of a functional immune system such as CD154 and CD80.52,53,54 Moreover, the microenvironment is potentially responsible for developing an immunosuppressive niche in the lymph nodes and marrow that allow for active immune evasion of the leukemic B-cells.55 Collectively, these cellular defects predispose patients to recurrent opportunistic infections especially with herpes zoster virus and cytomegalovirus (CMV).56,57 Defects in class switching of immunoglobulins and normal B-cell function also result in progressive hypogammaglobulinemia that predisposes patients to recurrent infections with encapsulated organisms.58 This may in part be related to the downregulation of CD154 on CLL B-cells or through CD95 interaction with its ligand.59,60 Understanding of these putative pathways has resulted in development of mechanistically relevant targeted therapy.61
ROLE OF THE B-CELL RECEPTOR PATHWAY
The B-cell receptor (BCR) plays an integral part in the development and maturation of B cells. Constitutive activation of the BCR is one of the most important survival signals for the propagation of CLL B cells.62 The surface immunoglobulin heterodimer that forms an integral part of the BCR, is critical for both antigen-dependent and antigen-independent signaling through the BCR.63,64,65 This signal is transduced through a variety of kinases including LYN (Lck/Yes novel), PI3K (phosphatidylinositol-4,5-bisphosphate 3-kinase), SYK (spleen tyrosine kinase), and BTK (Bruton tyrosine kinase).64 Their activation results in phosphorylation of phospholipase C-gamma-2 (PLCγ2) and induction of downstream second messengers that further modulate cell-survival regulators.35,63 Targeting the various kinases involved in the BCR pathway has resulted in significant improvements in the therapeutic options for this disease. Early results from studies done with BTK, PI3K, and SYK inhibitors have all shown excellent efficacy and tolerability, and these agents are currently being used in various combinations to improve disease outcomes.
BTK was initially characterized as a deficient kinase in patients with X-linked agammaglobulinemia, a disease characterized by a severe immunodeficient state.66 Specific mutations in BTK results in severe impairments in B-cell development and humoral immunity.66 Activating mutations of BTK have not been identified in CLL or other cancers. However, CLL B cells tend to have higher levels of BTK that can be induced through the BCR signaling pathway.67 Efficient targeting of BTK with the irreversible inhibitor ibrutinib results in significant abrogation of downstream survival signaling transduced through this pathway and results in the inhibition of cell survival and proliferation.67 Moreover, ibrutinib irreversibly targets interleukin (IL)-2–inducible T-cell kinase (ITK) in T cells, thus potentiating T-helper type 1 (Th1)–driven immune responses and reversing tumor-induced T-cell anergy.68
Similar to BTK, both SYK and PI3K can be induced by both the autonomous and antigen-dependent BCR activation and result in providing the critical signals that result in leukemic cell survival and proliferation.69,70 Similar to ibrutinib, the PI3K isoform delta inhibitor idelalisib antagonizes internal and external survival signals to the CLL cells and results in significant clinical response. Idelalisib is currently approved for the treatment of relapsed CLL.71 Similar results are observed with SYK inhibitors that are currently in early phase clinical trials.72
GENETICS OF CHRONIC LYMPHOCYTIC LEUKEMIA
Improved understanding of the genetics of CLL has resulted in significant improvements in our ability to determine the prognosis of this disease and to tailor therapy for our patients. Initial efforts to study the cytogenetic abnormalities in CLL were hindered by the inability of the tumor cells to proliferate in vitro for standard metaphase analyses. Improvements in our ability to stimulate the CLL B cells in vitro and the development of interphase fluorescent in situ hybridization techniques (FISH) have significantly improved and refined the study of cytogenetic abnormalities in this disease.73 Using these methods, del 13q14 was identified as the most common abnormality in patients with CLL, being present in approximately 50 percent of all patients, followed by trisomy 12, which is present in approximately 15 to 20 percent of patients, and del 11q22.3, which is present in approximately 10 to 15 percent of patients. Other abnormalities that were identified in significant numbers include del 6q21 and del 17p13.1. These abnormalities impart differential prognostic impact on disease outcomes.74
Functional studies have been performed to determine the association of these cytogenetic abnormalities to disease physiology. Specifically, deletion in the long arm of chromosome 13 results in a loss of the tumor-suppressor gene ARLTS1 with potential physiologic impact.75 Moreover, this region also includes genes encoding microRNA (miRNA) including miRNA-15 and miRNA-16.76 These abundant and evolutionarily conserved short, noncoding miRNA, that range in size from 21 to 25 nucleotides, have the potential to regulate the expression of a number of different genes at the posttranscriptional level significantly impacting cell signaling. They are also known to modulate the expression of various pro- and antiapoptotic proteins of significant relevance to CLL B-cell proliferation, including BCL-2 (miRNA-15 and miRNA-16), MCL-1 (miRNA-29), and TCL1 (miRNA-29 and miRNA-181).76,77,78 Recent studies have identified miRNA-150 as potentially the most abundantly expressed miRNA in patients with CLL and may be involved in the regulation of BCR signaling and subsequent survival signals.79 Multiple studies are currently underway to further define the full impact of the role of miRNAs in the pathogenesis of CLL and to develop strategies to modulate their expression and function for therapeutic benefit.
With regards to other cytogenetic abnormalities, trisomy 12 is often found in patients with progressive or relapsed disease or Richter transformation.80 Numerous genes are affected as a result of the trisomy and B cells obtained from patients with this abnormality tend to have higher surface expression of CD19, CD20, CD38, and immunoglobulins when compared to patients without this abnormality.80,81,82 Multiple genetic alterations have also been described in patients with del 11q22.3, but most importantly, it may involve the loss of the ataxia-telangiectasia mutated (ATM) gene, which normally activates p53 and results in either apoptosis or cellular repair in response to cytotoxic stimuli.83 Moreover, there is a decrease in the expression of miRNA-29 and miRNA-181, which are involved in the down regulation of the T-cell leukemia/lymphoma protein 1A (TCL1) oncogene.84 Patients with del 11q22.3 tend to have more aggressive disease with bulky lymphadenopathy85; historically have had a worse prognosis with disease poorly responsive to conventional nucleoside analogue-based therapy; and have specifically required the addition of alkylating agents like cyclophosphamide to overcome the poor prognostic impact.86,87
The del 17p13.1 is present in less than 10 percent of patients with CLL and frequently involves the deletion of the tumor protein p53 (TP53) gene that encodes the p53 protein which, as noted above, is critical for cellular apoptosis or repair specifically in response to cytotoxic stimuli.88 Patients with this abnormality have rapidly progressive disease with significantly inferior survival outcomes and poor response to therapy.89 This mutation also confers chemoresistance; consequently, responses to conventional chemotherapy-based regimens have been dismal.90 These patients also have higher risk of Richter transformation to more aggressive lymphomas over the course of their disease.89 The inferior outcomes observed in patients with del 11q or del 17p appears to persist in the era of treatment with kinase inhibitors, with patients experiencing a shorter progression-free interval, but are significantly better as compared to historic controls.91,92
Multiple other cytogenetic abnormalities have been reported from patients with CLL, including del 6q21, those involving the immunoglobulin heavy chain (IGH) locus on chromosome 14q32, and those involving the BCL-2 gene located on chromosome 18q21.93,94 These are generally associated with progressive, relapsed, and aggressive disease and frequently observed in patients with an unmutated immunoglobulin heavy-chain variable region (IGHV).93,95
Other recurring abnormalities in CLL involving SF3B1 (splicing factor 3B subunit 1), NOTCH1 (Notch homologue 1, translocation-associated), MYD88 (myeloid differentiation primary response gene 88), XPO1 (gene encoding exportin-1), KLHL6 (Kelch-like protein-6), and ERK1 (extracellular signal-regulated kinase 1) have been described using whole-exon and whole-genome sequencing with potential prognostic impact.96,97,98 With the advent of single nucleotide polymorphism (SNP) arrays, comparative genomic hybridization techniques, and determination of acquired copy number aberrations, more detailed studies can be performed assessing the mechanistic significance and impact of these genetic mutations on prognosis and customizing therapy.
CLINICAL PRESENTATION OF CHRONIC LYMPHOCYTIC LEUKEMIA
CLL is a disease of the elderly with the median age at diagnosis being 72 years. Most patients are asymptomatic at diagnosis and are diagnosed as a result of incidental finding of lymphadenopathy and lymphocytosis of uncertain etiology as part of an evaluation unrelated to CLL. A vast majority of patients may not have any significant symptoms related to the disease but some patients may experience mild fatigue or minor limitations in their activities of daily living. A subset of patients may present with recurring infectious complications, especially upper respiratory tract infections. Patients with advanced disease can uncommonly present with drenching night sweats, fevers, and weight loss (B symptoms), and signs and symptoms related to anemia, thrombocytopenia, and lymphadenopathy. The lymphadenopathy typically observed in patients with CLL is generally not fixed or tender and very rarely causes symptoms of organ dysfunction or resulting dependent-limb lymphedema. Patients can have exacerbations of their lymphadenopathy during an acute infectious episode, but this typically returns to baseline upon resolution of the underlying infectious complication.99 Splenomegaly is seen commonly in patients with CLL with resultant hypersplenism and thrombocytopenia. Significant hepatomegaly because of leukemic infiltration is unusual. CLL infiltration of multiple organs has been described but these are typically seen in patients with advanced disease and will occasionally cause symptoms. Pulmonary involvement has been observed in patients with high lymphocyte count and typically presents as an interstitial infiltrate on chest radiography. Chylous and hemorrhagic pleural effusions have also been reported.100,101,102 Similarly, leukemic infiltration of the gastrointestinal tract may result in chronic diarrhea or iron-deficiency anemia secondary to chronic bleeding or malabsorption. However, this mucosal infiltration is more commonly seen in patients with mantle cell lymphoma. CLL involvement of the central nervous system is rare and may result in headaches, confusion, meningismus, or cranial nerve palsies.103 More commonly, these patients are at higher risk for opportunistic infections of the central nervous system because of their deficient immune system. Patients with CLL are also known to have insect bite hypersensitivity.104,105 Patient’s typically present with recurrent, erythematous, painful eruptions usually on the exposed part of the extremities. Evaluation of skin biopsies from these patients reveal a mixed infiltrating population of T cells, B cells, and eosinophils. These resolve over time and can be effectively treated with a short course of glucocorticoid.
EVALUATION OF THE PATIENT WITH CHRONIC LYMPHOCYTIC LEUKEMIA
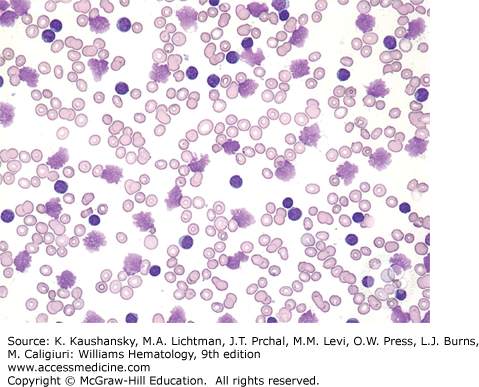
According to the IWCLL-2008 criteria, the diagnosis of CLL requires a sustained monoclonal lymphocytosis of greater than 5000 cells/μL of monoclonal B cells.1 This requires blood flow cytometry for immunophenotyping the B cells, which additionally reveals the cells to be positive for CD19, dim CD20, dim surface immunoglobulin, and negative for CD10, CD79b, and FMC7. A similar disease presentation with no evidence of hematopoietic involvement and with only lymph node involvement by cells of comparable morphology will be classified as small lymphocytic lymphoma. These patients are essentially managed similar to patients with CLL. CLL cells appear as small blue lymphocytes with scant cytoplasm on the Wright-Giemsa staining commonly used for evaluating blood films (Fig. 92–1). Smudge cells are also commonly observed on the blood film and this results from the mechanical disruption of the cells during the slide preparation process (Fig. 92–2). For an improved evaluation of the cellular morphology on the blood film, an albumin preparation is sometimes required. Patients can also have large prolymphocytes with prominent nucleoli in the blood but these lymphocytes must be less than 55 percent of the total lymphocyte population. The anemia is typically normocytic and normochromic and platelet morphology is typically preserved. A marrow aspirate and biopsy is not required for the vast majority of patients with CLL at initial presentation to establish a diagnosis. However, we do recommend performing a marrow aspirate and biopsy in patients with anemia and thrombocytopenia to evaluate the presence of autoimmune hemolytic anemia and/or immune thrombocytopenia. Marrow biopsy typically shows diffuse marrow involvement with a monotypic population of small lymphocytes. Variability of marrow involvement has been historically used as a potential marker for prognosis but has limited applicability given the availability of more specific and sensitive prognostic markers.106,107 The red cell precursors and megakaryocytes usually display an unremarkable morphology but do diminish in numbers with progressive disease. Conventional stimulated karyotype analysis and interphase FISH cytogenetic study, to evaluate for abnormalities commonly seen in patients with CLL, should be performed on all patients at the time of diagnosis and every time the disease changes character in order to determine the extent of a clonal evolution. Patients with atypical presentations, especially those with absent or low CD23 expression should have a negative FISH study for t(11;14) to exclude mantle cell lymphoma. Lymph node biopsy is not typically required for further establishing the diagnosis of CLL. Lymph nodes typically show architectural effacement by diffuse infiltration by cells of a similar morphology as observed in the peripheral circulation.
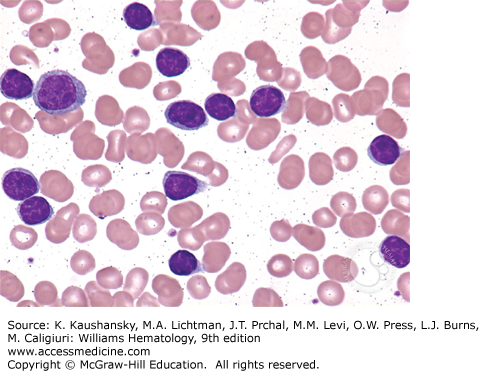
When anemia is present from CLL, patients typically will have normocytic and normochromic anemia, often with thrombocytopenia and lymphocytosis. Patients with a macrocytic anemia or an isolated anemia should have a Coombs test, haptoglobin test, and reticulocyte count performed to rule out autoimmune hemolytic anemia. These patients should also be evaluated for vitamin B12 and folic acid deficiencies, and malabsorption or gastrointestinal bleeding from CLL involvement should be ruled out.
Patients with CLL will frequently have hypogammaglobulinemia with decrease in the serum concentration of immunoglobulin (Ig) G, IgA, and IgM. The degree of hypogammaglobulinemia correlates with progressive disease and predisposes patients to recurrent sinopulmonary infections with encapsulated organisms. T-cell defects, which increase the risks of viral infections, have also been described; however, this is not routinely assessed at the time of initial presentation. A small percentage of patients also have monoclonal gammopathy with IgM or IgG or light-chain monoclonal paraproteinemia, which can be detected on serum protein electrophoresis and immunofixation. Excessively high heavy-chain paraproteinemia can result in symptoms related to hyperviscosity as seen in patients with Waldenström macroglobulinemia and should be managed as such.108,109,110 The presence of a monoclonal paraproteinemia and/or hypogammaglobulinemia may be related to inferior survival outcomes in patients with advanced disease stage, but not necessarily in patients with early stage disease.111,112
PROGNOSTIC MARKERS
All patients must undergo a comprehensive prognostic evaluation at the time of initial presentation. This allows the clinician to explain the specific disease characteristic to the patient and also helps the patient with the emotional adjustment process that they have to go through when initially diagnosed with this disease. All patients should undergo conventional karyotype analysis and stimulated interphase FISH either on blood or marrow aspirate. The minimum FISH panel should include assessment for del 17p13, del 11q23, trisomy 12, and del 13q14, and for t(11;14) in selected patients suspected of having mantle cell lymphoma. Conventional stimulated karyotype analysis is helpful in identifying the global structural abnormalities in chromosomes, especially of chromosomes 14, 3, and 6, that cannot be routinely detected on FISH analysis.113 Together, these assays have strong prognostic significance with regards to treatment-free and overall survival (Table 92–1).114 Patients with CLL acquire additional cytogenetic abnormalities, as detected by stimulated karyotyping and FISH analysis, with disease progression and especially after chemotherapy. This “clonal evolution” is predominantly observed in patients with unmutated IGHV and portends poor survival and inferior response to therapy.115 We therefore recommend repeating the stimulated karyotyping and FISH studies prior to initiation of a new line of treatment.
| Prognostic Variables | Median Survival (Months) | Median Time to First Treatment | |
|---|---|---|---|
| Interphase FISH cytogenetics | 13q– (sole) | 133 | 92 months |
| Trisomy 12 | 114 | 33 months | |
| Normal | 111 | 49 months | |
| 11q– | 79 | 13 months | |
| 17p– | 32 | 9 months | |
| IGHV mutational status | Unmutated (≥98 percent) | 89 | 3.5 years |
| Mutated (<98 percent) | >152 | 9.2 years |
The assessment of IGHV somatic mutation by a polymerase chain reaction–based assay has been shown to be an extremely reliable and important prognostic tool for patients with CLL. Patients with less than 2 percent homology in their nucleotide sequence as compared to consensus germline sequence are considered unmutated.116 Patients with a mutated IGHV, which is present in approximately 60 percent of patients with CLL, have a significantly prolonged treatment-free interval, longer remission durations, and overall survival (OS).117 These patients also have a very low incidence of clonal evolution or transformation to an aggressive histology.114,118 The IGHV mutation status does not vary over time and does serve as a reliable marker for predicting long-term disease outcomes.119 The only known exception to the mutation rule currently is the presence of IGHV 3–21 somatic mutation, which may confer an aggressive phenotype similar to leukemic cells from patients with unmutated IGHV at least in a subset of patients.120,121
Zeta-chain–associated protein kinase of 70 kDa (ZAP-70) is an intracellular tyrosine kinase that is typically associated with T-cell development and T-cell receptor (TCR) signaling. Expression of ZAP-70 in CLL B cells provides a survival advantage through intrinsic and extrinsic signals mediated through the BCR.122 Cytoplasmic assessment of ZAP-70 in CLL B cells by flow cytometry correlates strongly with IGHV mutational status and clinical outcomes, with an expression of 20 percent or more predictive of poor outcomes.123 The assessment of ZAP-70 by flow cytometric testing has been plagued with several issues, including lack of reproducibility and reliability of the reagents. Consequently, the National Comprehensive Cancer Network (NCCN) guidelines do not recommend the routine use of ZAP-70 as a prognostic marker outside of clinical trials. Given the stability of DNA and epigenetic modification by methylation, investigators have also sought to assess ZAP-70 expression by the absence of promoter methylation.124 Methylation analysis of select proximal 5′ regions of the ZAP-70 gene correlates very strongly with expression of ZAP-70 and has been established as an important and reliable prognostic marker with regards to predicting time to treatment and OS.125,126 This assay can be done by pyrosequencing with significant reproducibility among different laboratories.126
CD38 is a 45-kDa transmembrane glycoprotein that can be detected on the surface of CLL B cells by flow cytometry; a level of expression greater than 30 percent correlates strongly with progression-free survival (PFS).118 Newer reports, however, suggest that an even lower level of CD38 expression might also have a prognostic impact.127,128 CD38 appears to be involved in cellular metabolism by synthesizing cyclic adenosine diphosphate (cADP)-ribose from nicotinic acid adenine (NAD),129 and its activity and expression correlates with proliferation of the lymphocytes and progressive disease as demonstrated by their high Ki-67 proliferation index.130 CD38 expression also changes with disease progression and the assessment of the extent of expression of CD38 is based on nonstandardized, subjective parameters.131
CD49d can also be used as a reliable predictive marker. CD49d is a surface subunit of the integrin heterodimer that is involved in promoting survival of the CLL cells through growth signals derived from the microenvironment.132,133 Patients with 30 percent or greater cells expressing CD49d by flow cytometry are considered to be positive and constitute a group of patients with an aggressive disease course and inferior survival.134
Serum lactate dehydrogenase (LDH) and β2-microglobulin are readily available, validated markers of disease aggressiveness and prognosis. Specifically, β2-microglobulin is an independent prognostic marker for remission duration, PFS, and OS, and a higher level is observed in patients with advanced and extensive disease.135,136,137 Elevated LDH is associated with more-aggressive disease and with Richter syndrome. Lymphocyte doubling time could also be used as a tool to determine the prognosis of CLL. Patients with a lymphocyte doubling time of 12 months or less have worse OS and treatment-free survival.138 Other reasons for transient elevation in lymphocyte count should be ruled out before making the determination of lymphocyte doubling time. Thymidine kinase is an important intracellular enzyme; the soluble form can be detected in patients with CLL and predicts for advanced stage and progressive disease. However, the assay is not widely available and the test is rarely used in routine clinic care.139,140,141 Over the years, various other serum proteins have been found to be associated with various measures of disease outcomes, including soluble factors like CD23,142 CD44,143 vascular cell adhesion molecule (VCAM)-1,144 CD27,144 and matrix metalloproteinase (MMP)-9,145 IL-6,146 and IL-8.147 However, none of these is routinely used for clinical decision making (Table 92–2).
| Favorable Outcome | Unfavorable Outcome | |
|---|---|---|
| Lactate dehydrogenase | Low or normal | Elevated |
| Lymphocyte doubling time | >12 months | ≤12 months |
| Thymidine kinase activity | Low or normal | Elevated |
| β2-microglobulin | Low or normal | Elevated |
| Soluble CD23 levels | Low or normal | Elevated |
| CD38 expression | <30 percent | >30 percent |
| Interphase FISH cytogenetics | Normal Trisomy 12 13q– (sole) | 11q– 17p– |
| IGHV mutational status | Mutated (<98 percent) | Unmutated (≥98 percent) |
| CD49d expression | <30 percent | >30 percent |
Various miRNAs have also been validated as useful prognostic markers. miRNAs are noncoding RNAs that are 19 to 25 nucleotides in length and modulate mRNA translation and synthesis of various proteins. miRNA-15a and miRNA-16–1 were the first ones to be identified as underexpressed in CLL patient B cells.76 Genes for these miRNAs are located in the deleted region of chromosome 13q14 and modulate the expression of the antiapoptotic bcl-2 protein, which is overexpressed in patients with CLL and other B-cell lymphoproliferative disorders.148 Similarly, miRNA-34c is involved in patients with del 11q23 and regulates the expression of ZAP-70 and other proteins involved in the TP53 pathway.149 Using mass array methods, miRNA profiles have also been found to be predictive for disease progression, fludarabine resistance, and clinical outcomes.150,151
STAGING FOR CHRONIC LYMPHOCYTIC LEUKEMIA
The Rai152 and Binet153 staging systems have been used for a long time for patients with CLL (Tables 92–3 and 92–4). These easy-to-use staging systems are based on assessment of disease burden as determined by lymphadenopathy and splenomegaly demonstrated on physical examination and the presence of cytopenias. Further modifications and development of the Binet system established low-risk (stage 0: lymphocytosis only), intermediate-risk (stages 1–2: lymphocytosis with lymphadenopathy and hepatosplenomegaly), and high-risk groups of patients with CLL (stages 3–4: lymphocytosis with anemia and thrombocytopenia).153 These classification systems provided an estimate of median OS of 150, 90, and 19 months, respectively, and helped classify patients for subsequent therapeutic intervention. The staging systems still remain relevant and complementary to molecular testing in the modern era.154
| Stage at Diagnosis | Risk Level | Rai Stage at Diagnosis | Patients Never Requiring Therapy (%) | Median Survival (Months) |
|---|---|---|---|---|
| 0 | Low | Lymphocytosis >5 × 109/L only | 59 | 150 |
| 1 | Intermediate | Lymphocytosis + lymph node (LN) enlargement | 21 | 101 |
| 2 | Lymphocytosis + spleen/liver (S/L) enlargement ± LN | 23 | 71 | |
| 3 | High | Lymphocytosis + anemia (with hemoglobin <11 g/dL) ± LN or S/L | 5 | 19 |
| 4 | Lymphocytosis + thrombocytopenia (< 100 × 1012/L ± LN) or S/L | 0 | 19 |
| Stage at Diagnosis | Equivalent Rai staging | Rai Stage at Diagnosis | Proportion of Patients (%) | Median Survival (Years) |
|---|---|---|---|---|
| A | 0–2 | Lymphocytosis >5 × 109/L only with <3 enlarged nodal areas*; no anemia, no thrombocytopenia | 15 | 12+ |
| B | 1–2 | Lymphocytosis >5 × 109/L + ≥3 enlarged nodal areas*; no anemia, no thrombocytopenia | 30 | 7 |
| C | 3–4 | Lymphocytosis >5 × 109/L + anemia (hemoglobin <10 g/dL) or thrombocytopenia (<100 × 1012/L) regardless of the number of enlarged nodal areas* | 55 | 2 |
Marrow aspirate and biopsy is not routinely required for the initial diagnosis and management of the patient with CLL. Marrow biopsy is especially helpful in determining the etiology of thrombocytopenia and anemia, which can frequently be related to concomitant autoimmune processes. It may also be helpful in determining the extent of involvement by large prolymphocytes and in establishing the diagnosis of prolymphocytic leukemia. A diffuse pattern of involvement of the biopsy specimen by characteristic small lymphocytes is also associated with a worse prognosis than interstitial or nodular involvement.106,155 Ideally, a marrow biopsy is performed at the start of therapy to document the disease extent and to detect any atypical features.
Computed tomography (CT) scans are generally not required for the routine initial evaluation of patients with CLL and conventional staging systems rely on physical examination findings. Similarly, routine CT scans for the serial evaluation of disease extent have no role in patients with CLL. CT scans should be performed in patients with symptomatic disease and prior to starting therapy. Similarly, a positron emission tomography (PET) scan has no role in the routine management of patients with CLL. CLL lymph nodes are fluorodeoxyglucose (FDG) nonavid; however, FDG-PET scanning can be used for the identification of patients with Richter transformation with a high sensitivity and negative predictive value.156,157
Based on disease extent, characteristics and prognostic markers, various nomograms have been developed and validated for use in patients with CLL. These nomograms provide a robust predictive tool for outcomes and incorporates various aspects of the CLL patient and disease and can be used for standardized risk assessment.136,137,158
TREATMENT OF CHRONIC LYMPHOCYTIC LEUKEMIA
Treatment of patients with CLL is initiated at the time of symptomatic progressive disease. The specific criteria for initiating therapy have been detailed in the IWCLL-2008 guidelines.1 This recommendation is primarily based on older studies that failed to demonstrate a survival advantage in patients treated early in the course of disease.159,160 These results were validated by a large study of fludarabine treatment in patients with early stage disease conducted by the German CLL study group, which failed to show a survival advantage with early treatment using conventional chemotherapeutic agents.161 Trials are currently underway with kinase inhibitors to determine if early intervention can alter the natural history of the disease. We recommend initiating treatment when patients fulfill the IWCLL-2008 criteria for treatment, regardless of the prognostic factors.
Patients with autoimmune complications of CLL can be managed accordingly with steroids and immunosuppressive therapies prior to proceeding with definitive therapy for the underlying disease. Patients with CLL rarely exhibit evidence of leukostasis resulting from profound leukocytosis, therefore, an elevated white cell count should not be used as a sole criteria for initiating treatment. Similarly, hypogammaglobulinemia should not be used as a reason to treat the disease. Periodic intravenous immunoglobulin infusions can be used in patients with hypogammaglobulinemia and recurrent life-threatening infections with encapsulated organisms. The IWCLL-2008 criteria should also be used when determining the timing of therapy for patients with relapsed disease.
One of the most important factors to consider prior to initiating treatment is the functional state of the patient. Historically, age cutoff has been successfully used in developing specific therapies. Because the median age of diagnosis for CLL is 72 years, the vast majority of patients treated in the community are older and with multiple comorbid conditions. Unfortunately, there is limited data available in this patient population and until recently they had very limited options for therapy. Most of the clinical trial participants have been younger patients who are in their 50s and 60s. The gradual functional decline, decrease in organ function, especially renal and hepatic function, and an increase in the comorbid conditions in the majority of patients older than age 65 years significantly increases the risks and toxicities of conventional chemotherapeutic regimens especially nucleoside analogues. To address these issues, different approaches are being used to treat patients older than 65 years versus younger patients. A cumulative illness rating scale (CIRS) has been proposed and used primarily by the German CLL study group. It allows patients to be stratified based on an aggregate score derived from multiple factors including age, comorbid conditions and organ function.162
Chlorambucil has been used as the prominent alkylating agent for the treatment of CLL for the last 60 years. Chlorambucil is taken orally and is generally well-tolerated but does have multiple side effects, including nausea, vomiting, and cytopenias. Various doses and schedules have been used with different responses and tolerability profiles. Older studies compared chlorambucil to conventional high-grade lymphoma therapy, including cyclophosphamide, vincristine, and prednisone (CVP) and cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), and found no improvement in survival outcomes with the use of high-grade lymphoma-like therapies.159 Multiple doses of chlorambucil have been evaluated in various clinical trials with differing but similarly modest overall response rate (ORR) and PFS. No single-dosing regimen has been shown to be superior to another. One dosing regimen that has been used in cooperative group studies is 40 mg/m2 oral dose every 28 days for 12 cycles.163 An argument, however, can be made that escalating the dose of chlorambucil in younger patients might result in a higher response rate.164 Despite its ease of use and reasonable tolerability, chlorambucil is not very commonly used today because of the availability of better and potentially safer alternatives. Moreover, responses observed with chlorambucil are modest and not durable.165 Nevertheless, chlorambucil is potentially an option for treatment of elderly patients with multiple comorbidities and limited other options for treatment. It should, however, not be used for asymptomatic patients with early stage disease.160
Cyclophosphamide is also approved for the use in patients with CLL and has moderate efficacy and reasonable tolerability. Care should be taken to avoid nighttime dosing and aggressive hydration should encouraged to avoid hemorrhagic cystitis.166 Low-dose etoposide, either as a single agent or in combination with cladribine, also shows modest responses in patients with relapsed and refractory disease. Treatment with etoposide is associated with significant myelosuppression and resultant infectious complications.167,168
Bendamustine was approved for the treatment of patients with CLL in 2008. This was based on a phase III trial that demonstrated the superior efficacy of bendamustine as compared to chlorambucil with regards to response rate and PFS.169 Structurally, bendamustine has features common with alkylating agents and purine analogues, but its activity is primarily derived from the alkylating agent moiety.170 Bendamustine appears to be generally better tolerated than fludarabine but causes significant myelosuppression, requiring dose reductions, especially in elderly and infirm. Tolerability is better in patients with impaired renal function since excretion is primarily through the feces.171
Nucleoside analogues have also been used for the treatment of patients with CLL for the last 25 to 30 years. Fludarabine has been the most commonly used agent from this class of drugs. Fludarabine has moderate activity especially in the younger patients with good nutritional status and with untreated, early stage disease.172 After demonstrating promising activity as a single agent in early phase clinical trials, fludarabine was compared to chlorambucil in a randomized phase III study for the initial treatment of patients with CLL. Patients treated with fludarabine demonstrated improvement in PFS and OS compared to chlorambucil, albeit with a higher rate of infectious complications; however, all patients ultimately had recurrent disease.163 Similar to chlorambucil, fludarabine was also more effective compared to combination chemotherapy regimens like cyclophosphamide, doxorubicin, and prednisone (CAP).173 However, PFS and OS advantage was not shown in another randomized trial of elderly patients older than age 70 years treated with fludarabine on chlorambucil despite the higher overall and complete response (CR) rate in the fludarabine-treated patients.174
The use of fludarabine and similar nucleoside analogues is associated with significant hematologic and immunologic toxicities. Patients can experience prolonged cytopenias and especially neutropenia, which can result in a significant increase in the risk of infectious complications. These drugs are also exquisitely toxic to T cells, especially to CD4+ T cells. This effect may last for an extended period of time and predisposes patients to acquiring opportunistic infections.175,176 Neurologic toxicities have also been observed in patients receiving the usual dose of fludarabine.177 Patients treated with fludarabine occasionally develop autoimmune hemolytic anemia. In this situation, further use of fludarabine is contraindicated.178,179,180 Fludarabine is also poorly tolerated in patients with compromised renal function as a significant percentage of the metabolites are cleared via the renal system. Hemodialysis is a useful tool to limit fludarabine toxicity in patients who develop acute renal failure while receiving treatment.181 Moreover, fludarabine treatment in patients older than age 65 years was poorly tolerated and did not result in improvements in PFS and resulted in a trend toward inferior OS outcome when compared to chlorambucil.174
Other nucleoside analogues used in the treatment CLL include cladribine and pentostatin which have similar outcomes and toxicities as fludarabine.182,183,184,185 A phase III trial comparing fludarabine, cladribine, and chlorambucil showed similar ORR and CR rate with all three agents, but median PFS was superior with cladribine (25 months) versus 10 and 9 months with fludarabine and chlorambucil, respectively.186 No advantages, however, were observed in OS. Cytarabine has also shown modest activity, especially in combination with oxaliplatin, fludarabine, and rituximab (OFAR regimen) in patients with refractory disease and patients with Richter transformation.187,188
Multiple chemotherapeutic combinations have been studied in patients with CLL. One of the earlier combinations to be used was chlorambucil and prednisone with ORR of approximately 80 percent with CR rates of approximately 10 to 15 percent.165,189 This regimen was also found to be as effective as other combination regimens like CVP, cyclophosphamide, melphalan, and prednisone (CMP), CAP, or CHOP.159,190,191,192 A meta-analysis comparing lymphoma-like combination therapies found no improvements in survival outcomes over chlorambucil alone.159 High-dose combination therapies like CAP, CHOP, and CVP have no role in the routine management of patients with CLL.
Given the improvement in outcomes with single-agent fludarabine as compared to chlorambucil, multiple combinations were developed with fludarabine to improve on its efficacy. Both the combination of fludarabine and chlorambucil and fludarabine and prednisone were found to be similar to fludarabine and not developed further.172,193,194 Fludarabine was combined with cyclophosphamide (FC) and resulted in encouraging responses even in patients with heavily pretreated disease.195 Multiple phase III trials were subsequently conducted in predominantly younger patients, comparing fludarabine with FC and revealed an ORR of 74 to 94 percent with CR rates of 23 to 38 percent and median PFS of 33 to 48 months, all significantly better with the combination.87,196,197 Early toxicities were mostly related to cytopenias secondary to myelosuppression and the resultant increase in infectious complications resulted in a slight dose adjustment and a dose of fludarabine 25 to 30 mg/m2 for 3 days and cyclophosphamide 250 mg/m2 for 3 days every 28 days for six cycles was used for subsequent studies. Notably, the combination of FC was the first regimen that was able to overcome the adverse prognostic impact of del 11q.86
Similarly, other combinations of alkylating agents with nucleoside agents have been tested with varying success. These include cladribine and prednisone, which combination was shown to be better than chlorambucil and prednisone in terms of responses, but without improvements in OS and with a higher incidence of infectious complications in the cladribine-treated arm.198,199 Similarly, the addition of cyclophosphamide and prednisone to cladribine resulted in higher responses but more myelosuppression and related complications.200,201,202 Pentostatin was also combined with cyclophosphamide and resulted in ORR of 74 percent and CR rates of 17 percent in patients with fludarabine refractory disease.203 Pentostatin in combination with chlorambucil and prednisone also resulted in promising responses but was extremely immunosuppressive and resulted in an unacceptably high incidence of infectious complications.204
Fludarabine was also combined with mitoxantrone without significant improvements in outcome. Although mitoxantrone appeared to improve outcomes when added to cladribine, it came at the cost of significant toxicity.205,206 Likewise, the combination of fludarabine, cyclophosphamide, and mitoxantrone resulted in an ORR of 78 percent and CR rates of 50 percent in patients with relapsed disease and an ORR of 90 percent with a CR rate of 38 percent in patients with previously untreated disease. The major toxicity was myelosuppression.206,207
The advent of antibodies for the treatment of patients with CLL has been a major advance in the management of this disease with the first true consistent evidence of improving survival. Numerous antibodies targeting different receptors have been developed and are at various stages of development. Four antibodies are currently approved for routine management. Unfortunately, one of these (alemtuzumab) is no longer actively marketed for this indication.
Alemtuzumab is a CD52-targeting, humanized, monoclonal antibody that mediates its efficacy through direct cytotoxicity, complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC). CD52 is ubiquitously expressed on lymphocytes (including B and T cells) and monocytes and this explains the efficacy and toxicity of the antibody. Alemtuzumab is extremely effective in clearing the blood and marrow of disease and is also active in patients with del 17p disease which is generally refractory to conventional chemotherapy.208 However, alemtuzumab has limited efficacy in patients with bulky lymphadenopathy, especially in patients with lymph nodes that are greater than 5 cm in diameter.209,210,211 Alemtuzumab was initially approved in 2001 by the FDA for the treatment of patients who had failed prior therapy with nucleoside analogues. This was primarily based on small trials that administered alemtuzumab intravenously three times a week for 12 weeks and showed modest response rates of approximately 30 to 40 percent and CRs in less than 5 percent of patients.209,210,211 Responses were short-lived in this cohort of patients and the median response duration was approximately 9 months. Treatment was also complicated by infusion-related toxicities in the vast majority of patients. To minimize these reactions, alemtuzumab is started at a dose of 3 mg and escalated to 10 mg for the second dose and 30 mg for the third dose, as tolerated. Another major toxicity was immunosuppression that resulted in multiple recurrent infections especially with opportunistic organisms that are commonly seen in patients with chronic immunocompromised states like HIV/AIDS such as CMV, pneumocystis, or varicella zoster.209,210,211 Patients also experienced prolonged cytopenias, especially of natural killer (NK) and T cells, which can persist for more than 9 months following therapy.212 Consequently, antiviral and antimicrobial prophylaxis therapy should be initiated in all patients receiving alemtuzumab and should be continued for at least 6 months after completing therapy.
A subsequent large phase III (CAM307) clinical trial was performed that compared alemtuzumab to chlorambucil as first-line therapy.213 Two hundred ninety-seven patients were randomized to chlorambucil 40 mg/m2 every 4 weeks for 12 cycles or alemtuzumab 30 mg intravenous infusion three times per week for 12 weeks. Overall response with alemtuzumab was 83 percent, with 24 percent CRs and with time to next treatment of 23 months. This was significantly better than the results observed with chlorambucil, which resulted in an ORR of 55 percent with 2 percent CRs and time to next treatment of 14 months. Moreover, approximately one-third of patients treated with alemtuzumab achieved a minimal residual disease (MRD)-negative CR that was later shown to correlate with OS. Both agents were well-tolerated but alemtuzumab resulted in a higher incidence of CMV infections. This trial resulted in the approval of alemtuzumab as initial therapy for CLL in 2007.
Infusion reactions and infectious complications are the major issues observed with the use of alemtuzumab. Infusion reactions can be diminished with a subcutaneous administration which appears to be equally efficacious but with similar toxicity profiles.214,215 Despite the encouraging results and approvals by the FDA, alemtuzumab never gained mass popularity and was not used by practicing physicians very often. It is no longer being marketed by the company for CLL but (as of 2015) can be obtained upon written request at no cost. The limited future prospects of alemtuzumab precludes exhaustive discussion. Alemtuzumab has been studied in combination with chemotherapy and as consolidative therapy after chemotherapy and has shown some benefit, but is generally associated with significant serious infectious morbidity.216,217,218,219
Rituximab Rituximab is a chimeric, murine, CD20-targeting, monoclonal antibody that has been extensively used for the treatment of patients with CD20+ lymphoid malignancies. CD20 is a calcium channel that interacts with BCR complex and is ubiquitously expressed on B-cell non-Hodgkin lymphomas and has a weak expression on CLL cells. Rituximab exerts its efficacy through direct CDC and ADCC.220,221,222
The dose and schedule of treatment with rituximab was determined empirically and has since been modified repeatedly. Initial trials were performed with four weekly infusions at 375 mg/m2 and showed limited efficacy in patients with CLL.223,224 Responses were higher when higher doses (up to 2250 mg/m2) or dose-dense regimens (375 mg/m2 three times a week) were used but was primarily limited to the blood and nodal areas.225,226 Nonetheless, these studies established the efficacy of rituximab and supported combination trials with chemoimmunotherapy where their impact has been most impactful.
Rituximab is generally tolerated very well with the most common toxicity being infusion reactions that are predominantly observed primarily with the first dose. These are generally mild fevers or chills, but occasionally may result in serious reactions that mimic severe allergic or anaphylactic reactions or cytokine release syndrome. The infusion reactions can be minimized with the routine use of prophylactic acetaminophen, antihistamine, and corticosteroids, glucocorticoid, and by slowing the infusion rate. Patients may also experience transient, severe thrombocytopenia, the mechanism of which is poorly understood. Therefore, rituximab should be used with caution in patients with a preexisting thrombocytopenia. Another important and potentially severe toxicity is tumor lysis syndrome, which is generally observed in patients with a high circulating peripheral lymphocyte count. These patients should be monitored closely and should receive prophylactic hydration, allopurinol, and electrolyte monitoring during and after the infusion. Other uncommon toxicities include delayed neutropenia, hepatitis B reactivation, interstitial pneumonitis, rash, and serum sickness. Patients with a prior history of hepatitis B infection should receive monitoring for reactivation while being treated with rituximab or similar agents, with rapid implementation of antiviral therapy if reactivation is observed. Fatal cases of progressive multifocal leukoencephalopathy from JC polyomavirus infection has also been reported with the use of rituximab and similar monoclonal antibodies. These infections typically occur during treatment or soon after, with virtually all cases observed during the first year posttherapy.227,228
Ofatumumab Ofatumumab is a fully human, type 1, IgG1, CD20-targeting, monoclonal antibody that binds more effectively to a different epitope of CD20 than rituximab.229 In vitro it was shown to have improved CDC and ADCC as compared to rituximab.230,231 Ofatumumab is given as a test dose of 300 mg followed by eight weekly intravenous infusions of 2000 mg, after which patients can go on a maintenance schedule of four monthly infusions of 2000 mg. The half-life of ofatumumab is 21 days, but the B-cell–depleting effects may last for up to 7 months after the last infusion.232
Early results with ofatumumab as a single agent in patients who were refractory to alemtuzumab and/or fludarabine showed encouraging ORR of 58 percent in the double refractory group and 47 percent in the fludarabine refractory cohort with bulky disease. Responses were all partial except for one CR in the fludarabine refractory group. Response was also short lived and the median duration of response was 7 months in the fludarabine refractory group and 5.6 months in the double refractory group with most patients progressing during treatment.233 These results led to the approval of ofatumumab in patients with relapsed disease refractory to alemtuzumab and/or fludarabine in 2009. Subsequent studies compared ofatumumab (300 mg on day 1 followed by eight weekly infusions of 1000 mg, followed by 1000 mg on day 1 of subsequent 28-day cycles, for a maximum of 12 cycles) given in combination with oral chlorambucil (10 mg/m2 on days 1 to 7 of each 28-day cycle) versus chlorambucil alone, in patients with previously untreated CLL who required treatment and were not considered candidates for conventional chemoimmunotherapy. The combination resulted in an ORR of 82 percent versus 68 percent with chlorambucil alone. However, CR rates were 12 percent versus 1 percent and median duration of response was 22 months versus 13 months in the combination versus chlorambucil arms, respectively.234 Based on these results, the combination of ofatumumab and chlorambucil was granted approval in 2014 for the treatment of previously untreated patients with CLL for whom fludarabine-based therapy is considered inappropriate.
Ofatumumab has also been studied in the upfront setting in combination with FC in smaller phase II studies of a relatively young patient population (median age: 56 years). This study revealed an ORR of 75 percent with 41 percent CRs.235 The combination of ofatumumab with pentostatin and cyclophosphamide resulted in a 96 percent ORR with 46 percent complete remission rate.236,237 This compares favorably to the results seen with the combination of rituximab and chemotherapy, but large multiinstitution randomized trials are lacking.
Ofatumumab is generally well-tolerated, with the most common reaction being an infusion-related reaction that typically occurs with the first infusion and includes fevers, rash, fatigue, chills, and diaphoresis. These reactions tend to get better with subsequent infusions. Infectious complications are similar to those reported with other CD20 monoclonal antibodies.
Obinutuzumab Obinutuzumab is a CD20-targeting antibody that was approved in combination with chlorambucil for the initial treatment of patients with CLL in 2014. Obinutuzumab is a fully humanized, type II, IgG1 antibody238 with additional structural modifications that explain its enhanced activity. It binds selectively to the extracellular domain of CD20 with reduced internalization. This persistence of the antibody on the cell surface along with a fucosylation in its Fc region allow for enhanced ADCC through robust engagement of Fc-gamma receptor type III on effector cells. Another modification in the hinge region allows for more potent direct cytotoxicity.239,240,241 Together, these modifications translate into a higher efficacy as compared with rituximab in both preclinical and clinical studies.239,240,241,242,243
Obinutuzumab in combination with chlorambucil was compared to rituximab and chlorambucil and chlorambucil alone in patients with untreated CLL in the CLL-11 trial conducted by the German CLL study group.244 Patients had a median age of 73 years, which is closer to the median age of 72 years at diagnosis for CLL patients, and significantly higher than the median ages of 58 to 62 years which have historically been the population that has been enrolled in chemotherapy-based clinical trials for CLL. More importantly, these patients had clinically meaningful comorbid conditions. Treatment of this patient population has historically been challenging and no prior chemotherapeutic option, including fludarabine, has improved survival outcomes as compared to chlorambucil alone.174 The combination of obinutuzumab and chlorambucil improved ORRs (77.3 percent [obinutuzumab and chlorambucil] vs. 65.7 percent [rituximab and chlorambucil] vs. 31.4 percent [chlorambucil]), CR rates (22.3 percent [obinutuzumab and chlorambucil] vs. 7.3 percent [rituximab and chlorambucil] vs. 0 percent [chlorambucil]) and median PFS (26.7 months [obinutuzumab and chlorambucil] vs. 16.3 months [rituximab and chlorambucil] vs. 11.1 months [chlorambucil]). Obinutuzumab with chlorambucil also prolonged OS over that which was observed with chlorambucil. Notably, the combination also resulted in significant improvement in the rate of MRD-negative status in both marrow (19.5 percent vs. 2.6 percent) and blood (37.7 percent vs. 3.3 percent) as compared to rituximab and chlorambucil.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


