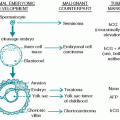
I. EPIDEMIOLOGY AND ETIOLOGY
A. Incidence. Chronic lymphocytic leukemia (CLL) is the most common type of leukemia in Western countries, accounting for one-third of cases. The incidence in the United States is 3.5 per 100,000. The disease is rare in Asians. Ninety percent of patients are >50 years of age, and the median age at diagnosis is approximately 65 years. Men are affected more often than women by a ratio of 2:1.
B. Etiology
1. Genetic factors. The vast majority of cases are sporadic, but familial clusters of CLL have been described. The incidence in relatives of patients with leukemia is two- to threefold greater than that of the general population. The etiology in the majority of cases is unknown.
2. Immunologic factors. Inherited and acquired immune deficiency is often associated with CLL and other lymphoproliferative neoplasms. This observation supports a concept that defective immune surveillance may result in proliferation of malignant cell clones and increased susceptibility to potential leukemogenic transduction, such as by viruses.
3. Molecular and cytogenetic aberrations. Somatic mutations of the immunoglobulin gene take place in the germinal center of secondary lymphoid follicles after antigen exposure. IgVH hypermutations are detected in approximately half of the cases of CLL, indicating that the cells are derived from postgerminal center or memory B cells and do not express ZAP-70, a molecule usually required for selective activation of T cells but aberrantly expressed in some cases of (B-cell) CLL. Some cases of CLL show characteristics of naive B cells with unmutated antigen receptors and are ZAP-70 positive.
Chromosomal abnormalities are detected in >80% of cases of CLL by use of interphase fluorescence
in situ hybridization (FISH). CLL is characterized by loss or gain of chromosomal genetic material rather than by translocations. Conventional cytogenetics typically miss these abnormalities. Incidence, genes involved, and clinical features of the most common chromosome abnormalities are shown in
Table 23.1.
4. Radiation and cytotoxic agents. Populations exposed to ionizing radiation or cytotoxic chemotherapy are not associated with an increased incidence of CLL.
II. PATHOLOGY AND NATURAL HISTORY
A. Pathology. CLL is characterized by suppression of programmed cell death (apoptosis) of mature B cells. Increased levels of Bcl-2 protein have been identified in cells taken from patients with CLL. Two other
BCL-2 family gene products may prevent apoptosis of CLL cells.
BCL-xL, which is expressed at high levels in CLL, enhances the efforts of
Bcl-2, whereas
BCL-xS, which is expressed at low levels in CLL cells, is a
Bcl-2 antagonist. Furthermore, nonclonal
CD4+ T cells and cells of the bone marrow stroma may support and sustain survival of the neoplastic B-cell clone.
1. The leukemia cells have low levels of surface immunoglobulin and display a single heavy chain class, typically µ; some cells display both µ and δ; and less commonly, γ, α, or no heavy chain determinant is found. The leukemia cells display either κ or λ light chains, but never both.
2. Surface membrane antigens include the B-cell antigens CD19, CD20, and CD23; CD22 and CD79b are weak or negative. The CD11c and CD25 antigens are found on the cells in half of the cases. CD5 is always present on CLL cells. Expression of CD38 has been associated with an unfavorable prognosis (see
Appendix C5, “Discriminatory Immunophenotypes for Lymphocytic Neoplasms”).
3. CLL is associated with low expression of the B-cell receptor (BCR), leading to impaired responses of CLL cells after BCR signaling.
B. Natural history
1. Immunologic abnormalities in CLL
a. Advanced disease is associated with hypogammaglobulinemia and an increased risk of infection with encapsulated bacterial organisms.
b. A variety of in vitro lymphocyte function tests are abnormal. Many studies have suggested decreased helper T-cell functions, and patients may have an inversion of the normal helper T-cell-to-suppressor T-cell ratios.
c. Monoclonal paraproteins are not routinely identified; however, when one uses more sensitive techniques, it appears that most patients with CLL secrete small amounts of paraproteins (usually immunoglobulin M [IgM]). These paraproteins rarely produce symptoms of hyperviscosity.
d. Coombs-positive warm antibody hemolytic anemia occurs in about 10% of patients and immune thrombocytopenia in about 5%. Immune neutropenia and pure red blood cell aplasia are rare.
e. Compared with the general population, the incidence of skin carcinoma is increased eightfold and visceral epithelial cancers twofold in patients with CLL.
2. Clinical course. The natural history of CLL is highly variable. Survival is closely correlated with the stage of disease at the time of diagnosis. Because most patients are elderly, >30% die of diseases unrelated to leukemia.
a. Manifestations. In 25% of patients, CLL is first recognized at routine physical examination or by a routine CBC. Clinical manifestations
develop as the leukemia cells accumulate in the lymph nodes, spleen, liver, and bone marrow.
(1) Pulmonary infiltrates and pleural effusions are common late in the course of disease.
(2) Renal involvement is common in CLL, but functional impairment is unusual in the absence of obstructive uropathy, pyelonephritis, or hyperuricemia secondary to tumor lysis from therapy.
(3) Transformation into a diffuse large cell lymphoma (Richter syndrome) or prolymphocytic leukemia occurs in <5% of patients.
(4) Skin involvement is rare.
(5) Osteolytic lesions and isolated mediastinal involvement are unusual and suggest a diagnosis other than CLL.
b. Progressive disease is accompanied by deterioration of both humoral and cell-mediated immunity. As the disease progresses, patients develop progressive pancytopenia, persistent fever, and inanition. During the latter stages of disease, cytotoxic chemotherapy is generally ineffective, and dosages are restricted because of pancytopenia. Death is usually caused by infection, bleeding, or other complications of the disease.
(1) Herpes zoster is the cause of 10% of infections in CLL patients.
(2) Bacterial pathogens associated with hypogammaglobulinemia include Streptococcus pneumoniae, Haemophilus influenzae, and Legionella sp.
(3) Pneumocystis jiroveci may be the causative infectious agent in patients with pulmonary infiltrates.
IV. STAGING SYSTEM AND PROGNOSTIC FACTORS
A. Prognostic factors. Routine CBC may detect asymptomatic cases of CLL, but this has no bearing on the overall survival of these patients. If survival has been improved, effective treatment of complicating infections in CLL probably has been responsible for much of the improvement.
1. Clinical staging is helpful for determining prognosis and deciding when to initiate treatment. Anemia and thrombocytopenia adversely affect prognosis when they are due to leukemic infiltration (“packing”) of the bone marrow but not when they are due to autoimmune destruction of red blood cells or platelets.
2. The pattern of bone marrow infiltration also appears to affect prognosis. Patients with nodular or interstitial patterns of bone marrow involvement have longer survival than patients with diffuse (“packed”) involvement.
3. V genes. Two subsets of CLL are defined by the IgVH mutational status. Patients with somatic mutations of the VH genes generally have a better prognosis than those with unmutated VH genes.
4. CD38 expression on CLL cells generally is associated with a poorer prognosis than absent or low-level expression of CD38.
5. Chromosome abnormalities as described in
Table 23.1 may predict outcome.
6. Other adverse prognostic factors appear to be a lymphocyte doubling time of <12 months and an elevated serum β2-microglobulin level.
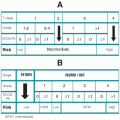
B. Staging system. The
modified Rai classification of CLL with median survivals is shown in
Table 23.2 (see Section III.C for differences with the NCI Working Group criteria).
V. MANAGEMENT
A. Indications for treatment. CLL is usually indolent. Treatment of asymptomatic stable disease is not warranted. The magnitude of the blood lymphocyte count does not indicate the need to start therapy. The initiation of therapy should be timed according to the clinically assessed pace of disease. Complete remission is not a necessary goal. The indications for instituting therapy in CLL are as follows:
1. Persistent or progressive systemic symptoms (fever, sweats, weight loss)
2. Lymphadenopathy that causes mechanical obstruction or bothersome cosmetic deformities
3. Progressive enlargement of the lymph nodes, liver, or spleen
4. Stage III or IV (high-risk) disease that results from the replacement of bone marrow with lymphocytes
5. Immune hemolysis or immune thrombocytopenia that is treated with prednisone alone
6. Rapid lymphocyte doubling time
B. Chemotherapy. Fludarabine is superior to alkylating agents in its associated complete response rate and duration of response but not in overall survival. Drug dosage schedules for CLL are as follows:
1. Nucleosides. Fludarabine may be the initial treatment of choice for patients who would benefit from a rapid and sustained remission, such as those designated for further aggressive therapy. Prolonged treatment with fludarabine and other nucleoside analogs, such as cladribine or pentostatin, however, are also associated with marked immunosuppression and an increased risk of opportunistic infections and autoimmune hemolysis, and may be associated with myelodysplasia.
a. Fludarabine, 25 to 30 mg/m2 IV daily for 5 consecutive days every 4 weeks
b. Cladribine (2-chlorodeoxyadenosine, 2-CdA), either 0.10 mg/kg daily by continuous IV infusion for 7 days, or 0.14 mg/kg daily IV over 2 hours for 5 consecutive days every 4 to 5 weeks
c. Pentostatin at 4 mg/m2 IV every 2 weeks. Typically, this drug, and the others mentioned above, are combined with an alkylating agent and/or rituximab.
2. Alkylating agents remain useful and effective for palliative therapy.
a. Chlorambucil, 0.1 mg/kg PO daily for 3 to 6 weeks as tolerated; the dose is usually tapered to 2 mg daily until the desired effect is achieved. Alternatively, 15 to 30 mg/m2 PO may be given for 1 day (or divided over 4 days) every 14 to 21 days; the dose is adjusted to tolerance.
b. Cyclophosphamide, 2 to 4 mg/kg PO daily for 10 days; the dose is then adjusted downward for continued therapy until the desired effect is achieved.
c. Bendamustine is a bifunctional alkylating agent with antimetabolite properties. Doses of 100 mg/m2 IV are given over 30 minutes on days 1 and 2 every 28 days for up to 6 cycles. The dose is adjusted for cytopenias.
3. Monoclonal antibodies can be useful for CLL.
a. Rituximab (Rituxan) is an anti-CD20 chimeric monoclonal antibody. The dose of 375 mg/m
2 weekly for 4 weeks, as used for non-Hodgkin
lymphoma, has minimal activity in previously treated patients with CLL but is quite useful as part of combination therapy in untreated patients. Dose escalation with the weekly schedule or thrice-weekly administration increases the clinical response significantly with minimal toxicity. Unlike alemtuzumab, the remission rate is low. However, rituximab is not associated with myelosuppression and is a better candidate, in terms of immunosuppression, to combine with chemotherapy than alemtuzumab. The primary toxicity associated with rituximab is an infusion-related cytokine release syndrome that is typically associated with the first infusion.
b. Alemtuzumab (Campath-1H) is a humanized anti-CD52 monoclonal antibody whose antigen is expressed on more than 95% of mature B and T lymphocytes and may be used for the treatment of fludarabinerefractory CLL. Alemtuzumab preferentially eliminates CLL cells from the blood, bone marrow, and spleen but is less effective in nodal sites of disease. Approximately one-third of the patients will have a partial response to alemtuzumab; complete responses are rare. Dosing is discussed in
Chapter 4, Section VII.B.
Side effects of alemtuzumab include cytokine release syndrome, immunosuppression, and neutropenia. The acute infusion reactions following intravenous administration are markedly reduced with subcutaneous injection. The immunosuppression has resulted opportunistic infections; trimethoprim/sulfamethoxazole (Bactrim) and acyclovir are recommended for prophylaxis.
4. Modern combination therapies have resulted in high response rates (70% to 95%) and high complete response rates (20% to 65%) in previously untreated patients. Variations in the following regimens are in active clinical trials. Prophylaxis with fluconazole, acyclovir, and Bactrim are recommended for all of these therapies.
a. Fludarabine and cyclophosphamide. Fludarabine (25 mg/m2 IV daily on days 1 to 3) and cyclophosphamide (250 mg/m2 IV daily on days 1 to 3) are given every 4 weeks for six cycles (30% to 50% achieve a complete remission, CR).
b. Fludarabine and rituximab. Fludarabine (25 mg/m2 IV daily on days 1 to 5) is given every 4 weeks for six cycles. Rituximab (375 mg/m2) is given on days 1 and 4 of the first cycle and on day 1 of cycles 2 to 6 (50% CR rate).
c. Fludarabine, cyclophosphamide, and rituximab. Fludarabine (25 mg/m2 IV daily on days 1 to 3) and cyclophosphamide (250 mg/m2 IV daily on days 1 to 3) are given every 4 weeks for six cycles. Rituximab is given at a dose of 375 mg/m2 1 day before the first course and increased to 500 mg/m2 on day 1 for cycles 2 to 6 (65% CR rate).
d. Pentostatin and cyclophosphamide. Pentostatin (4 mg/m2 IV) and cyclophosphamide (600 to 900 mg/m2 IV) are given every 3 weeks for six cycles (17% CR in previously treated patients). A newer regimen with rituximab uses lower doses of pentostatin (4 mg/m2) and cyclophosphamide (600 mg/m2).
e. Bendamustine and rituximab. Bendamustine (70 mg/m2 IV over 30 minutes on days 1 and 2) and rituximab (375 mg/m2 for the first course and 500 mg/m2 for subsequent courses). Repeat every 28 days.
5. Treatment of
resistant disease is controversial. Clearly, if patients were initially treated with an alkylating agent, then fludarabine or a fludarabine combination (see above) should be initiated. If a patient is resistant to fludarabine, then single-agent alkylators, alemtuzumab, or pentostatin
plus cyclophosphamide should be considered. However, if patients have previously responded to fludarabine, then one of the fludarabine combinations should be considered. The role of autologous and allogeneic stem cell transplants is limited in CLL patients, who are typically elderly and poor candidates for transplantation. However, in selected patients, nonmyeloablative allogeneic stem cell transplants can be considered to induce long-term remission.
C. Radiation therapy (RT). Local irradiation is recommended only for reduction of lymph node masses that threaten vital organ function and that respond poorly to chemotherapy. Splenic irradiation may result in improvement of disease elsewhere and may temporarily improve signs of hypersplenism; however, the clinical usefulness of splenic irradiation has not been established. Total-body irradiation remains investigational and potentially dangerous.
D. Surgery. Splenectomy is indicated in CLL patients who have immune hemolytic anemia or immune thrombocytopenia that either fails to respond to corticosteroid therapy or must be treated with corticosteroids chronically. Splenectomy may also be helpful in patients with problematic hypersplenism.