Chemotherapy for Pediatric Solid Tumors
Barbara A. Gruner
Pediatric cancers are rare, with an incidence of 169.1 per 1,000,000 for children under the age of 19 in the United States annually.1 Cancer is the leading cause of disease-related death in children in the same age group,2 with solid tumors representing approximately two-thirds of the new childhood cancer diagnoses each year. In this chapter, current chemotherapy regimens are reviewed for four of the more common tumors in this age group: neuroblastoma, Wilms tumor (WT), rhabdomyosarcoma, and retinoblastoma. Together, these tumors represent about one-fifth of new cancer diagnoses in children and are most commonly seen in the youngest patients. Important factors influencing the outcome for these patients include patient’s age at diagnosis, genetics, extent of the tumor at diagnosis, and response to therapy. New developments in chemotherapy regimens through single-institution and cooperative group clinical trials have led to improvements in outcomes over the past several decades.
NEUROBLASTOMA
Neuroblastoma is the most common extracranial malignancy in children representing 8% to 10% of all childhood tumors.3 Approximately 600 new cases are diagnosed each year with an annual incidence in the United States of 8.2 per 1,000,000 under age 19.1,3 The majority of cases are diagnosed in children under age 5 and 98% of cases are seen in children under age 10.4 In the United States, there is a slightly higher incidence in boys than in girls and in whites than in blacks. Tumors may occur anywhere along the sympathetic chain, although the most common site is the adrenal gland. Because it may appear at an early age, screening for neuroblastoma has been investigated in several countries with the hope of identifying high-risk patients at a point when treatment would be most effective. In Japan, which had a mass screening program for children at 6 months, investigators found that they were not identifying the patients with advanced (i.e., high-risk) disease.4 Screening programs in other countries have yielded similar results.5
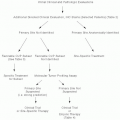
Neuroblastoma behaves differently depending on the age of the patient at diagnosis and the stage (i.e., extent) of disease. The tumor has been known to regress spontaneously and to be noted only as an incidental finding on chest X-rays or other imaging done for other reasons. Infants with metastatic disease respond well to treatment, whereas the outcome for older children with metastatic disease is poor. Genetic factors and morphologic features play an important part in determining prognosis in neuroblastoma. The tumor’s DNA index, the level of expression of the MYCN gene, or the presence of chromosome 1p and/or 17q deletion4,6 are associated with prognosis, as are more recently identified prognostic markers, such as trkA gene expression, which is inversely related to MYCN gene expression.7 Expression of other genetic markers that are independent of MYCN expression is being investigated.8,9 The most widely accepted histopathologic classification system for neuroblastoma is the International Neuroblastoma Pathology Classification (INPC) system, also referred to as the Shimada system.10 This INPC system combined the original Shimada classification11 with modifications made by Joshi12,13 and is the classification system presently used by the Children’s Oncology Group (COG).
Staging evaluation for a child with neuroblastoma involves CT or MRI of the primary site and of potential metastatic sites, chest X-ray, bilateral bone marrow aspirations and core biopsies, and metaiodobenzylguanidine scan. The tumor markers that are best in monitoring disease response are urine homovanillic acid and vanillylmandelic acid. Open biopsy is required to provide diagnostic tissue and to grossly resect the tumor or to reduce tumor burden, if possible. Core-needle biopsy may be preferable to an open procedure if the tumor is thought to be unresectable.14 The International Neuroblastoma Staging System (INSS)14 is now nearly universally accepted for assigning stage in neuroblastoma. Briefly, stage 1 tumor is localized with gross total resection; stage 2, tumor with gross residual after resection; stage 3, unresectable tumor, which crosses the midline; stage 4, metastatic tumor; and stage 4S, localized tumor with metastases in bone marrow, liver, or skin in a patient <365 days old. In COG studies, the patient is assigned to a prognostic risk group based on the patient’s age, INSS stage, MYCN status, Shimada histology, and tumor cell DNA index.
Low-risk Neuroblastoma
Children in the low-risk group based on prognostic stage are generally cured with relatively modest therapy. Apparently, tumors that have low MYCN expression and favorable histology more often are sensitive to therapy.15 In stage 1 disease, surgery alone produces a survival rate of over 90% regardless of age, histology, MYCN status, or DNA index.16 The majority of patients with stage 2 disease should be treated with surgery alone. However, patients who are symptomatic at diagnosis or have evidence of recurrent or progressive disease should receive chemotherapy. Recommended chemotherapy consists of cisplatin, etoposide, cyclophosphamide, and doxorubicin for four to eight monthly cycles, depending on the tumor response. Only patients older than 1 year at diagnosis whose tumors are
MYCN amplified and have unfavorable histology should not be considered low risk. The presence of these features would place the patient in the high-risk group.
MYCN amplified and have unfavorable histology should not be considered low risk. The presence of these features would place the patient in the high-risk group.
Intermediate-risk Neuroblastoma
The intermediate-risk group patients are those with (1) INSS stage 3, <1 year of age, with tumor of any histology or DNA index, but without MYCN amplification, or are those with (2) INSS stage 3, older than 1 year with tumor with favorable histology without MYCN amplification, or are those with (3) INSS stage 4, <1 year of age at diagnosis, with tumor of any histology or DNA index without MYCN amplification. Following their initial surgery, these patients are treated with combination chemotherapy, consisting of carboplatin, etoposide, cyclophosphamide, and doxorubicin for four to eight cycles. The length of chemotherapy depends upon histology and tumor response. Surgery is used to remove residual tumor if present at the end of the chemotherapy. The event-free survival (EFS) was 88% at 3 years. For patients with stage 3 disease, the EFS was 92%, and for patients with stage 4 disease, 81%.17
High-risk Neuroblastoma
The high-risk neuroblastoma patients have INSS stage 3 or 4 disease, are of any age, and have tumor of unfavorable histology and/or tumor with MYCN amplification. Recommended treatment includes intensive multiagent chemotherapy, followed by autologous stem cell rescue (ASCR)18 and involved field irradiation. Continuation therapy consists of cis-retinoic acid.19 In randomized trials,18,19 consolidation with ASCR was directly compared with consolidation with chemotherapy. In each case, estimated EFS at 5 years18 and 3 years19 with ASCR was superior to chemotherapy alone, that is, 38% to 27%, and 34% to 22%, respectively. There does not appear to be a significant difference in the outcome between patients treated with purged19 or unpurged18 autologous marrow. In the randomization between continuation therapy with cis-retinoic acid and no further therapy, the outcome with cis-retinoic acid was superior, 46% versus 29% 3-year EFS. Overall, the EFS for patients who received ASCR and cis-retinoic acid was superior to those who received chemotherapy alone. A recent randomized COG phase III trial compared continuation with cis-retinoic acid or with immunotherapy consisting of anti-GD2 antibody, interleukin-2, granulocyte-macrophage colony-stimulating factor, and cis-retinoic acid in patients with high-risk neuroblastoma. The patients who received immunotherapy had an improved EFS (66% vs. 46% at 2 years) compared with those who received cis-retinoic alone.20
A study from the French Society of Pediatric Oncology also compared high-dose chemotherapy with ASCR and conventional chemotherapy in a small number of patients with INSS stage 2 or 3 neuroblastoma and tumor with MYCN amplification.21 EFS for patients in the ASCR group was 83% at 5 years versus 25% for children who received conventional chemotherapy (stage 2 patients, 100% vs. 0% [n = 8], and for the stage 3 patients, 80% vs. 38% [n = 23]). The number of relapses in the ASCR group was also less than in the chemotherapy group.
Recurrent Neuroblastoma
Survival remains poor for children with recurrent neuroblastoma in spite of the advances in frontline therapy. A retrospective review of 31 cases at the Hospital for Sick Children,22 showed that, in the majority of cases, tumor relapses occurred within the first 24 months after diagnosis (n = 24), and that 28 of the patients who relapsed died of progressive disease. The factors that led to poor outcome in relapse were the same ones that were associated with aggressive disease at diagnosis: age at diagnosis, INSS stage 4 disease, unfavorable histology, MYCN amplification, and chromosome 1p deletion. Regimens containing topotecan and cyclophosphamide have shown promise for patients in first relapse.23,24,25
WILMS TUMOR
WT is the second most common abdominal tumor and the most common renal tumor in children. The incidence is 6 cases per 1,000,000 annually in children below age 19 years.1 WT represents 6% of all pediatric tumors, making it the second most common extracranial solid tumor diagnosis.26 It is associated with mutations in the WT1 gene located on the short arm of chromosome 11, at the 11p13 locus, in both sporadic and hereditary cases. Syndromes in which WT1 mutations are found include Denys-Drash syndrome and the WAGR (WT aniridia, genitourinary abnormalities, and mental retardation) syndrome.26,27 A second tumor suppressor gene, WT2, has been identified in WT. WT2 mutations are found on the short arm of chromosome 11, at the 11p15 locus. This gene is associated with overgrowth syndromes such as Beckwith-Wiedemann syndrome and hemihypertrophy.26,27 Other genetic alterations include those in the long arm of chromosome 16, in the short arm of chromosome 1, in the short arm of chromosome 7, and in the p53 gene.27 It should be noted that the vast majority of WTs occur in otherwise healthy children. Clinical presentation may include abdominal or flank mass, abdominal pain, hematuria, fever, and hypertension.
The dramatic improvements in outcome for children with WT in the last several decades are because of the development of effective multimodal therapy evaluated rapidly in large, multicenter clinical trials conducted by the National Wilms Tumor Study Group (NWTSG) in the United States and Canada and the International Society of Pediatric Oncology (SIOP) in Europe. The primary difference between the approaches taken by the two study groups is the timing of surgery. The SIOP trials employ preoperative chemotherapy without histologic confirmation of WT,28 whereas the NWTSG trials generally utilize primary surgery.29 In the trials from both study groups, experimental choice of the postoperative chemotherapeutic regimen depends on tumor stage and histology. Tumor staging is done at the time of surgery and is similar in both groups: stage I is completely resected local tumor, stage II is tumor with local extension but completely resected, stage III is unresectable tumor, stage IV is metastatic tumor, and stage V is bilateral tumor at diagnosis. The primary difference in staging is the designation of stage II N0 and stage
II N1 in the SIOP system, which falls under NTWSG stages II and III, respectively.
II N1 in the SIOP system, which falls under NTWSG stages II and III, respectively.
Histologic subtyping for WT depends on the presence or absence of anaplasia. Tumor cells that are large and hyperchromic with large nuclei that have bizarre, multipolar mitotic figures are classified as anaplastic.26 The absence of anaplasia indicates a favorable prognosis (favorable histology), whereas the presence of tumor anaplasia indicates unfavorable prognosis (unfavorable histology). If the anaplasia is widespread or diffuse rather than focal, the tumor is considered higher risk and the patient requires more intensive therapy.30
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


