Chemistry and Biosynthesis of Thyrotropin
Ronald N. Cohen
Fredric E. Wondisford
Thyrotropin (thyroid-stimulating hormone, TSH) is the main regulator of thyroid hormone biosynthesis and secretion. TSH reference limits increase with age, and vary somewhat between racial and/or ethnic groups (1). Although daily TSH levels can exhibit some fluctuations, e.g., due to normal diurnal rhythms or from abnormal situations ranging from acute sleep deficiency (2) to severe illness, typical variations of thyroid hormone and TSH levels for each given individual are generally small, suggesting a unique set point (3). Therefore, TSH availability and actions are critical determinants of thyroid function.
Chemistry
TSH is one of the four related glycoprotein hormones synthesized either by the anterior lobe of the pituitary gland or the placenta. TSH, pituitary luteinizing hormone (LH), pituitary follicle-stimulating hormone (FSH), and chorionic gonadotropin (CG) consist of two non-covalently linked α- and β-subunits (4). The amino acid sequence of the α-subunit is common to all four of these hormones within any mammalian species. The β-subunit of each hormone has a distinct amino acid sequence, and this subunit carries the specific information relating to receptor binding and hormonal activity. Free β-subunits are devoid of bioactivity, though, and require non-covalent combination with the common α-subunit to express this information. Some recent evidence suggests that β-subunits provide binding specificity, whereas α-subunits may be involved in receptor activation (5).
Thyroid-stimulating activity has been extracted from virtually all mammalian pituitary glands, as well as from those of lower vertebrates. Important chemical information has been derived from studies of bovine, porcine, and human TSH (4). TSH preparations purified from pituitary glands by various chromatographic procedures contain heterogeneous but closely related components, which have variable biologic activities. These components can be separated by gel electrophoresis, isoelectric focusing, or chromatofocusing. The molecular weights of the components of mammalian TSH are in the range of 28,000 to 30,000 daltons. Differences in molecular weight are attributed to heterogeneity of the oligosaccharide chains, heterogeneity at the amino terminus, and the extent of amidation of glutamic and aspartic acid residues.
The human α-subunit contains a protein core of 92 amino acid residues, while the bovine α-subunit contains 96 amino acids. The α-subunits contain 10 half-cystine residues, all of which are in disulfide linkage, as are those in the β-subunits. Knowledge of the specific location of the disulfide bonds in both subunits has been advanced by x-ray crystallographic analysis of the closely related glycoprotein, human CG (6,7), and the three-dimensional structure of TSH likely contains the cystine-knot motif and basic folding patterns found in human CG (8).
The β-subunits dissociated from intact human TSH have a protein core of 112 amino acids; however, the complementary DNA for human TSH-β predicts a protein of 118 amino acids, 6 more than that present in the isolated protein. When the linear sequences of β-subunits from different species are aligned to juxtapose half-cystine residues (12 in number), many regions of similar or identical sequences are apparent. From such relationships, and the fact that each can recombine with the common α-subunit, it seems probable that the three-dimensional structures of different β-subunits are similar. Indeed, evidence from chemical modification studies indicate that the regions around amino acid residues 51 to 57 and 75 to 80 of the β-subunit are involved in interaction with the common α-subunit. Based on the similarities in sequences among the β-subunits, all probably originated from a single ancestral gene.
Both the α- and β-subunits of TSH, as well as of LH, FSH, and CG, contain covalently linked carbohydrate chains. For TSH, LH, and FSH, these chains are linked to asparagine residues (N-linked), and for the β-subunit of human CG there are additional linkages to serine residues (O-linked). Moreover, the free α-subunit that is secreted also contains an additional site of O-glycosylation (9). In human TSH, the sugar residues include mannose, fucose, N-acetylglucosamine, galactose, N-acetylgalactosamine, and sialic acid. TSH and LH also contain an unusual sulfate group that terminates certain chains; such sulfation is found only to a small extent in FSH, and not at all in CG (10,11). The sugar residues are three oligosaccharide units, all of which are heterogeneous and whose specific structures vary with developmental and endocrine states (12). Recombinant human TSH has been produced in large amounts (see later in the chapter), and the biologic properties of particular isoforms of TSH and clinical use of TSH are being investigated (13,14,15,16). More recently, a CHO cell line expressing rat α2,6-sialyltransferase was used to create a human-like sialylated recombinant TSH molecule (17).
Although extrapituitary sources of TSH or TSH subunit variants have been suggested, their in vivo significance remains unclear (18,19,20).
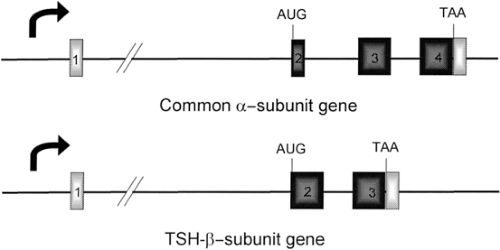 Figure 10A.1 Schematic representation of the genes for the subunits of human thyrotropin (TSH). The genes for the α- and β-subunits have four and three exons (numerals), respectively, represented by boxes separated by introns (thin lines). The coding exons are denoted by black boxes, and the untranslated regions are represented by white boxes. The translation start (AUG) and stop (TAA) codons are shown in their relative position below each gene; the start of transcription is denoted by a bent arrow. |
Genes Encoding the Subunits of Thyrotropin
Chromosome Localization
The gene encoding the common α-subunit of human CG, FSH, LH, and TSH, and the genes encoding their respective
β-subunits are each located on different chromosomes. The location of the common α-subunit and specific β-subunit on different chromosomes raises interesting questions about how their expression patterns are coordinately regulated during hormone synthesis. Moreover, the gene for the β-subunit of CG is the only β-subunit gene of this family that exists in more than one copy; at least two of the seven copies on chromosome 19 are actively transcribed.
β-subunits are each located on different chromosomes. The location of the common α-subunit and specific β-subunit on different chromosomes raises interesting questions about how their expression patterns are coordinately regulated during hormone synthesis. Moreover, the gene for the β-subunit of CG is the only β-subunit gene of this family that exists in more than one copy; at least two of the seven copies on chromosome 19 are actively transcribed.
Structure of the Common α-subunit Gene
The α-subunit gene has been isolated from a variety of species, including cows (21), mice (22), rats (23), and humans (24). The organization of each gene is similar in that it contains four exons and three introns, and all are of approximately the same size. The human gene is 9.4 kilobases (kb) in length and contains three introns of 6.4 kb, 1.7 kb, and 0.4 kb. Intron 1 is located between the 5′-untranslated region contained in exons 1 and 2; intron 2 interrupts the α-subunit peptide-coding region in codon 6; and intron 3 is between codons 67 and 68 of the peptide (Fig. 10A.1). In rats and cows, intron 2 interrupts the peptide-coding region within amino acid 10, resulting in a mature α-subunit peptide that is four amino acids longer than the human α-subunit peptide (96 amino acids vs. 92 amino acids).
A single transcription start site has been found in each of these genes by mapping studies of pituitary RNA. Upstream of these start sites are consensus TATA boxes, thought to be important for correct and efficient transcription by RNA polymerase II (25). In the human α-subunit gene, the TATA box is 26 base pairs (bp) from the transcription start site (24). In addition, there is considerable homology among these species in other 5′-flanking regions, including a palindromic sequence of TGACGTCA in the human α-subunit gene that confers responsiveness to cyclic adenosine monophosphate (cyclic AMP) (26,27). However, the palindrome is altered in the other species to TGATGTCA; at least in cows, this change reduces its cyclic AMP responsiveness dramatically (28).
Structure of the Thyrotropin-β–subunit Gene
The TSH-β–subunit gene has been isolated from rats (29), humans (30,31,32), and mice (33,34). The rat and human genes contain three exons. The mouse gene contains five exons: The two additional exons are 5′-untranslated regions; these are unique due to changes in the sequence of the gene in mice (30). With this exception, the organization of these genes is quite similar in that there are 5′-untranslated exons separated from the TSH-β–coding region by a large first intron (Fig. 10A.1). The first exon of the human TSH-β gene is 37 bp and is untranslated, while the second exon encodes the leader peptide and first 34 amino acids of the mature TSH-β peptide; the third exon contains the remaining coding region (amino acids 35 to 118) and 3′-untranslated sequences.
The start of transcription has been determined in the rat, mouse, and human TSH-β genes. Both the rat and mouse genes contain two transcriptional start sites approximately 40 base pairs (bp) apart, as assessed by primer extension or S1 nuclease analysis of pituitary RNA (29,33,34). Most transcription initiates from the downstream site (90% to 99%), and both sites are preceded by consensus TATA box sequences. Transcription from the downstream site is dramatically increased in hypothyroid rats and mice, while transcription from the upstream site is either unaffected or reduced by thyroid hormone (29,33,34).
The TSH-β gene in humans contains only one transcriptional start site in a location similar to that of the downstream site in the TSH-β gene in rats and mice (30,31,32). This difference may be due to an alteration in the upstream TATA box, which is changed from TATATAA in the rat and mouse gene to TGTATAA in the human gene. There may be an additional start site in the human gene (35). However, based on RNA mapping studies done in one TSH-secreting adenoma, this site does not correspond to the upstream TATA box in the human TSH-β gene.
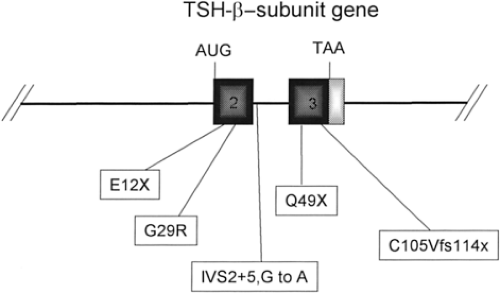 Figure 10A.2 Point mutations of the human thyrotropin (TSH)-β–subunit gene in patients with familial TSH deficiency. Three point mutations in this gene have been described (see text). Two of these mutations (E12X and G29R) occur in exon 2, and predict either a truncated TSH-β–subunit or one with an amino acid change in the CAGY region. The single letters define the wild-type amino acid (preceding the number of the amino acid) and the substituted amino acid (after the number of the amino acid); fs, frame shift. |
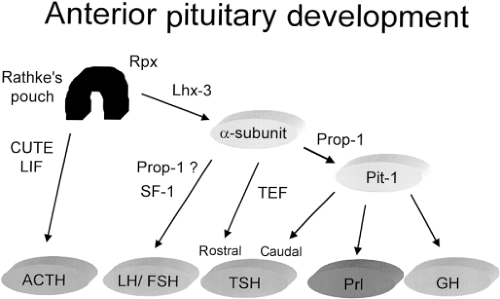 Figure 10A.3 Schematic representation of development of the anterior pituitary gland. The sequential expression of transcription factors leads to cell-specific expression of pituitary hormones. ACTH, corticotropin; CUTE, corticotroph upstream transcription binding protein; GH, growth hormone; Prl, prolactin; Prop-1, Prophet of Pit-1; SF-1, steroidogenic factor-1; TEF, thyrotroph embryonic factor. |
Central Hypothyroidism Due to Thyrotropin-β–subunit Gene Abnormalities or Alterations in Thyrotroph Development
Several kindreds with central hypothyroidism and undetectable or at least inappropriately low serum TSH concentrations due to mutations in the TSH-β–subunit gene have been described (36,37). Two affected members of a Japanese family had a point mutation in exon 2 that changed a glycine residue to an arginine residue (G29R) (36). This mutation is in a region that is highly conserved among glycoprotein subunit genes (CAGY)
and is thought to be important in subunit combination (Fig. 10A.2). This mutation results in synthesis of a TSH-β–subunit that cannot associate with the α-subunit, and therefore no TSH heterodimer is produced.
and is thought to be important in subunit combination (Fig. 10A.2). This mutation results in synthesis of a TSH-β–subunit that cannot associate with the α-subunit, and therefore no TSH heterodimer is produced.
Two related Greek families have been described in which affected family members had a mutation in the β-subunit of TSH that causes premature termination of synthesis of the subunit (E12X) (37). A Turkish family was found to have a different mutation that also results in premature termination of synthesis of the subunit (Q49X) (38). In another family a mutation was found in the second intron of the β-subunit gene (G29R) that predicts skipping of exon 2 and generation of a 25 amino acid nonsense peptide from an aberrant start site in exon 3 (39). A Brazilian kindred with central hypothyroidism was described in which there is a frame-shift deletion in codon 105 (C105Vfs114X) of the β-subunit gene (40). This mutation substantially reduces TSH secretion, although some TSH can be detected in the serum of affected patients. Based on the crystallographic structure of the related hormone, CG, the mutation interferes with a disulfide bond that stabilizes the αβ heterodimer of TSH (6). This mutation may be the most frequent β-subunit mutation (41). All of these disorders are autosomal recessive in inheritance, and affected family members do not produce TSH with biologic activity (Fig. 10A.2).
Central hypothyroidism can also be caused by defects in pituitary development, which results in impaired formation or survival of thyrotrophs and thus expression of TSH. In general, pituitary organogenesis is characterized by the orderly expression of cell-specific transcription factors (Fig. 10A.3), including Pit-1, Prophet of Pit-1 (Prop-1), P-Lim (Lhx-3), Ptx, Hesx-1 (Rpx), and thyrotroph embryonic factor (TEF) (for review see reference 42).
TEF is expressed in the region of developing thyrotrophs, at the time of onset of expression of the β-subunit of TSH (43). TEF binds to sites in the promoter region of the β-subunit gene to regulate expression of the gene. In contrast, Pit-1 is expressed after the initiation of expression of the TSH-β–subunit gene (44). However, Pit-1 is required for thyrotroph survival and further expression of the TSH-β–subunit gene. While there are Pit-1–dependent and Pit-1–independent thyrotrophs in the developing anterior pituitary, the latter population disappears before birth (44,45). Pit-1 also regulates the expression of growth hormone and prolactin, and is required for normal somatotroph and lactotroph function. In mice, Pit-1 mutations result in TSH, growth hormone, and prolactin deficiency and pituitary hypoplasia (46). Pit-1 mutations in humans cause analogous defects, a syndrome termed combined pituitary hormone deficiency (CPHD) (Fig. 10A.4) (47,48). A well-characterized Pit-1 mutation implicated in hypopituitarism in humans is the R271W mutation (49), which leads to production of a mutant Pit-1 that binds DNA but does not transactivate appropriately. These patients have hypothyroidism, but may have inappropriately normal serum TSH concentrations. They have a blunted serum TSH response to thyrotropin-releasing hormone (TRH), suggesting the importance of Pit-1 in the TRH signaling pathway.
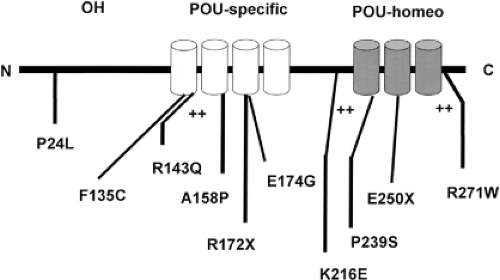 Figure 10A.4 Point mutations in the human Pit-1 gene in patients with combined pituitary hormone deficiency, as defined using the single-letter code for amino acids. POU-specific and POU-homeodomains, and the corresponding α-helices, are outlined in relation to indicated mutations in the Pit-1 gene. ++, lysine- and arginine-rich domains; N, the amino terminus of the Pit-1 protein; C, the carboxyl terminus of the Pit-1 protein. |
Prop-1 gene expression precedes the expression of Pit-1, and appears to be required for the development of Pit-1–dependent cell lines. Ames mice, which have hormonal defects similar to those in Snell and Jackson dwarf mice, have a mutation in the alpha helix of the homeodomain of the Prop-1 gene (50). Similar to Snell and Jackson dwarf mice, analogous defects have been found in some patients with CPHD (51,52,53). These patients have central hypothyroidism as well as growth hormone and prolactin deficiency, similar to patients with Pit-1 mutations. However, patients with mutations in Prop-1 also tend to exhibit LH and FSH deficiency. One common Prop-1 mutation is a 2-bp deletion (A301G302) (53), which results in impaired DNA binding (51).
Pre-Translational Regulation of Thyrotropin Biosynthesis
Negative Regulators
Thyroid Hormone
The major negative regulator of TSH biosynthesis is thyroid hormone as part of the feedback system of the hypothalamic–pituitary–thyroid axis. Thyroid hormones exert their effects by binding to thyroid hormone receptors (TR), cellular homologs of the viral erythroblastic leukemia oncogene (v-erbA). TRs are derived from two separate gene loci in mammals: α and β (54). The α locus in humans is located on chromosome 17 (17q11.2) and the β locus on chromosome 3 (3p24.3). The TR-α gene undergoes alternative splicing, generating two main isoforms: TR-α1 and TR-α2. TR-α1 is a bona fide TR, whereas alternative splicing of the C-terminus of TR-α2 generates a non-triiodothyronine (T3)–binding isoform. The TR-β gene generates two major isoforms due to alternative promoter utilization: TR-β1 and TR-β2. This results in two β receptors, each with a different amino terminus. Recent evidence suggests that a number of additional short TR isoforms lacking important functional domains may also exist, but their roles in vivo remain uncertain (55).
TRs are variably expressed in mammals. In general, TR-α1 and TR-β1 are ubiquitously expressed. TR-α1 is most highly expressed in the brain and heart, whereas TR-β1 is most highly expressed in the liver. In contrast, TR-β2 expression is limited principally to the hypothalamus, pituitary, cochlea, and retina (56). Differences in the expression patterns as well as structure of TRs suggest that one or both of these characteristics may determine thyroid hormone action in particular tissues.
TRs are members of the nuclear hormone receptor superfamily. TRs share in common with other members of this superfamily a conserved DNA-binding region, which contains two coordinated zinc molecules. Carboxy terminal to the DNA-binding domain is the ligand-binding domain, which determines ligand-binding specificity. This domain is not highly conserved among different members of this superfamily. Similarly, the amino terminus domain, N-terminal to the DNA-binding domain, differs significantly between receptors. In addition to known ligands for members of this superfamily, other receptors known as orphan receptors have been described for which no ligands have yet been identified.
TRs bind to DNA as a homodimer or a heterodimer with the retinoid X receptor, also known as RXR (57). TR homodimers dissociate after binding T3, whereas TR–RXR heterodimers do not. This has led to the suggestion that TR–RXR heterodimers are the most important determinants of T3 action. The molecular mechanisms underlying thyroid hormone gene stimulation have been fairly well elucidated, though negative regulation by thyroid hormone is still not understood (see later). The TR binds to the so-called “positive response elements” on genes whose expression is stimulated by thyroid hormone. On these response elements, the TR recruits additional nuclear cofactors. In the absence of T3, TRs bind a class of proteins termed nuclear corepressors. These proteins in turn recruit histone deacetylase (HDAC) enzymes to the transcription complex, which results in histone deacetylation and increased chromatin packing (58). When T3 binds to its receptors, the nuclear corepressors are released and nuclear co-activators are recruited. These include the p160 (or SRC) class of co-activators, the co-integrator class of cofactors (CBP and p300), and the Mediator complex. Certain of these proteins exhibit intrinsic histone acetylase activity, which results in histone acetylation and unwrapping of the chromatin. Presumably, this configuration of chromatin allows easier access to RNA polymerase II, and transcription is increased. In contrast, Mediator interacts directly with the RNA Polymerase II complex.
Thyroid hormone inhibits the synthesis and secretion of TSH at the level of the pituitary and indirectly at the level of the hypothalamus by reducing the secretion of TRH (59,60,61,62). In animals, thyroid hormone administration results in a dramatic decrease in the levels of messenger RNA (mRNA) for the common α- and TSH-β-subunits due to a reduction in transcription of the respective genes (59,60). However, the magnitude and rapidity of suppression varies among the subunits. In mouse TSH-secreting tumor cells exposed to T3, the level of TSH-β mRNA falls more rapidly (50% inhibition at 4 hours) and to a greater extent (>95% suppression at 4 hours) than that of the common α-subunit. In addition, after prolonged T3 exposure, TSH-β–subunit mRNA was undetectable, whereas the level of α-subunit mRNA remains at approximately 25% of control levels (59).
In addition to the expected suppression of TSH secretion by thyroid hormone, paradoxical increases in TSH secretion
have been noted soon after initiation of thyroid hormone replacement, followed by later suppression of TSH secretion. In one study, administration of a low dose of T3 to patients with hypothyroidism resulted in an increase in the serum TSH response to TRH (63). This effect may be due to a generalized defect in protein synthesis, corrected by low doses of T3.
have been noted soon after initiation of thyroid hormone replacement, followed by later suppression of TSH secretion. In one study, administration of a low dose of T3 to patients with hypothyroidism resulted in an increase in the serum TSH response to TRH (63). This effect may be due to a generalized defect in protein synthesis, corrected by low doses of T3.
As opposed to the discussion of corepressors and co-activators above (for genes whose expression is stimulated by thyroid hormone), the mechanisms underlying negative regulation of the TRH and TSH-subunit genes by thyroid hormone are not clear. The thyroid hormone–inhibitory elements in these genes (the so-called “negative response elements”) have unique properties, as compared with thyroid hormone–stimulatory elements (for review see reference 64). First, certain of the inhibitory elements in these genes do not bind RXRs, and, unlike stimulating TR elements, RXRs antagonize TR binding to these elements (65). An exception is an important negative thyroid hormone response element in the TRH gene promoter that binds TR monomers, TR homodimers, and TR–RXR heterodimers (66). Although some models suggest that negative regulation by thyroid hormone can occur in the absence of TR binding to DNA, in vitro and in vivo studies have demonstrated that this binding is required for thyroid hormone inhibition (67,68).
Studies in mice in which one or several of the TR isoforms were deleted have revealed that TR isoforms differentially regulate the production of TSH (69,70,71,72,73). The most important isoform controlling TSH production is TR-β2 (71). In the hypothalamus, it is perhaps the only isoform responsible for negative regulation of the TRH gene (74). It is also the most important isoform for negative regulation in the pituitary, although the TR-α1 and TR-β1 isoforms play some role at the extremes of serum thyroid hormone concentrations. Mice deficient in p160 co-activators display variable resistance to thyroid hormone in the pituitary (75,76,77,78), and it has been shown that an intact co-activator binding surface is absolutely required for proper negative regulation by thyroid hormone (79). In contrast, mice expressing a mutant form of NCoR that cannot be recruited to the TR complex display normal TSH levels in the setting of reduced thyroid hormone (80).
Steroid Hormones
TSH-subunit gene expression is altered by steroid hormones, including glucocorticoids, estrogen, and testosterone. In humans, high doses of dexamethasone decrease serum TSH concentrations (81); dexamethasone also decreases TSH secretion in patients with TSH-secreting pituitary adenomas. In a study of mice with thyrotropic tumors, dexamethasone decreased TSH secretion, but the levels of TSH-subunit mRNA in the tumor did not change (82). Therefore, dexamethasone may exert its effect on TSH-subunit biosynthesis at a translational or posttranslational level. Of note, patients with untreated Cushing’s syndrome also exhibit diminished TSH secretion, which increases after surgical cure of the disease (83).
In animals and humans, estrogen administration does not alter basal or TRH-stimulated serum TSH concentrations (84). In hypothyroid rats, however, high doses of estradiol augment thyroid hormone–stimulated suppression of synthesis of α- and β-subunit mRNA (84). In animals, testosterone has similar effects, perhaps caused by its peripheral conversion to estrogen. In contrast, progesterone has been hypothesized to enhance the inhibitory effect of sleep on TSH secretion (85). In sum, steroid hormones are not major regulators of TSH secretion under normal conditions, but they may play a role during some pathologic conditions such as hypothyroidism.
Related posts:
 Anatomy and Pathology of the Thyrotrophs
Anatomy and Pathology of the Thyrotrophs
 Iodine Deficiency and Endemic Cretinism
Iodine Deficiency and Endemic Cretinism
 Toxic Adenoma and Toxic Multinodular Goiter
Toxic Adenoma and Toxic Multinodular Goiter
 Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
 Overview of the Clinical Manifestations of Hypothyroidism
Overview of the Clinical Manifestations of Hypothyroidism
 Hypothyroidism in Infants and Children: Neonatal Screening
Hypothyroidism in Infants and Children: Neonatal Screening
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


