Abstract
The newborn screening (NBS) program is a well-established comprehensive public health initiative with the main goal of identifying newborns affected by genetic disorders, for whom early interventions may prevent disease morbidity and mortality. The early-in-life screening for genetic conditions not only permits early institution of specific therapeutic measures for those affected, but also creates the opportunity for genetic counselling for carriers (e.g., parents). Several hematologic conditions have benefited from NBS, most notably hemoglobinopathies, particularly sickle cell disease (SCD), for which early diagnosis with preemptive penicillin initiation has substantively reduced pediatric mortality [1, 2].
Introduction
The newborn screening (NBS) program is a well-established comprehensive public health initiative with the main goal of identifying newborns affected by genetic disorders, for whom early interventions may prevent disease morbidity and mortality. The early-in-life screening for genetic conditions not only permits early institution of specific therapeutic measures for those affected, but also creates the opportunity for genetic counselling for carriers (e.g., parents). Several hematologic conditions have benefited from NBS, most notably hemoglobinopathies, particularly sickle cell disease (SCD), for which early diagnosis with preemptive penicillin initiation has substantively reduced pediatric mortality [1, 2]. The inclusion of severe combined immune deficiency (SCID) in the panel of screened genetic disorders has allowed for early referral to hematopoietic stem cell transplantation and the soon-to-be scaled up, gene therapy [3, 4].
Principles of a NBS program should include informed consent, use of accurate laboratory methods, provision of relevant education and counseling, procurement and immediate care institution, preferably within the comprehensive care programs. In addition, NBS should be voluntary, every effort made to protect patient and family confidentiality, and avoid labeling and stigmatization.
In the United States (US), the number of genetic conditions screened at birth varies by state, however all 50 states screen for hemoglobinopathies. Severe combined immunodeficiency (SCID), although not a primary hematologic genetic condition, may present with hematologic symptoms at birth (e.g., lymphopenia), and has been part of the recommended universal newborn screening panel (RUSP) since 2010. Currently, all states in the US include SCID in their NBS panel. On the other hand, only the NBS programs of Pennsylvania and District of Columbia screen for glucose-6 phosphate dehydrogenase (G6PD) deficiency. The next subsections will discuss the NBS efforts for quantitative and qualitative hemoglobinopathies, and for G6PD deficiency which is the most common red blood cell (RBC) enzymopathy. In addition, the efforts to screen and treat different forms of SCID have also been described.
NBS Implementation for Blood Disorders in the United States and Worldwide
The first legislatively mandated state NBS program in the US was established in 1963, when Massachusetts began testing dried blood specimens (DBS) obtained from newborns heel prick for phenylketonuria (PKU), using the Guthrie test [5]. PKU testing was eventually implemented nationwide by 1969–1970. By the mid 1970s many states had expanded their screening panel to include other metabolic (maple syrup disease, homocystinuria), and endocrine (congenital hypothyroidism) disorders [6, 7]. New York was the first to include testing for SCD on its NBS panel in 1975. The solubility tube test to screen for hemoglobin S (HbS) has been known since 1949 [8]. However, despite the refinement (by using a more stable reducing agent than dithionite) and automation of this technique, it was limited because it was unable to distinguish among homozygous (HbSS), trait (HbAS), and compound heterozygous (e.g., HbSC) HbS mutations [9–11]. The development of cellulose acetate electrophoresis technique in 1969 to separate Hb variants, and its eventual adaptation to test elutes from DBS in 1973, enabled large-scale screening for SCD in conjunction with PKU testing using the same specimen [12, 13]. Since then, techniques such as isoelectric focusing (IEF) and high performance liquid chromatography (HPLC) have increasingly been utilized by NBS state programs [14]. Introduction of tandem mass spectrometry (MS/MS) technology in the 1990s revolutionized the field as it enabled rapid, cost effective, and highly sensitive testing for multiple simultaneous metabolic disorders using a single assay [15]. Its application has recently been extended to screen for clinically relevant Hb variants [16]. In 1972, a centralized Hemoglobinopathy Reference Laboratory was established at the Centers for Disease Control and Prevention (CDC) to formulate protocols, standardize laboratory techniques, and perform regular evaluation to ensure regulated quality controlled testing for hemoglobinopathies nationwide [17, 18]. After its closure in 1993, this role was transitioned to the Newborn Screening Quality Assurance Program at the CDC, to ensure the continued quality and accuracy of testing provided by NBS laboratories nationwide [19, 20].
Two NHLBI-sponsored studies, in 1977 and 1983, namely the Co-operative Study of Sickle Cell Disease (CSSD) and Prophylactic Penicillin Study, respectively, demonstrated that penicillin prophylaxis started in infancy significantly decreased invasive pneumococcal infection and associated mortality [1, 21]. Based on these findings, the NIH consensus conference in 1987 emphasized the importance of early identification of SCD by universal NBS so as to prevent fatal complications during infancy [22]. Despite this recommendation, it was only in 2007 that screening for SCD was adopted by all 50 states [23]. In 2005, the American College of Medical Genetics (ACMG) was commissioned to give recommendations for a universal screening panel (RUSP) [24]. This panel identified 29 disorders (20 metabolic and 9 non-metabolic), including SCD, that comprised the core screening panel for NBS programs in the US. These disorders were selected based on the following criteria: (1) they could be identified in pre-symptomatic newborns 24–48 hours after birth, (2) their natural history was well understood, (3) validated screening tests with appropriate sensitivity and specificity existed, and (4) effective treatment to mitigate the disease-associated adverse outcomes were available. G6PD screening was excluded from this panel due to insufficient knowledge of the natural history of the G6PD mutations [25]. An additional 25 disorders that did not fulfil the above criteria, but could be incidentally detected during the screening for the core panel conditions, were considered secondary/target conditions, and the state programs were then required to report their findings. Since then, the ACMG has continued to expand the RUSP. In November 2016, the RUSP included 34 core and 26 secondary conditions, however, its implementation into state NBS programs has not been uniform. This is due to the rarity of these conditions, heterogeneity in their clinical presentation and disease course, and the lack of established infrastructure required to provide high-quality medical services for afflicted children, raising questions regarding the cost-effectiveness and benefit of their early screening [26–27].
Since the implementation of NBS for hemoglobinopathies in the US, other countries have adopted it. In low and middle income countries (LMICs), NBS coverage depends on federal financing and prioritization. Other sources of funding include private laboratories, which offer screening on a fee-for-service basis, and collaboration among local non-government agencies and the World Health Organization regional offices. Except for Brazil, Panama, Costa Rica, and Cuba [28], the NBS coverage for hemoglobinopathies in LMICs is variable [29]. In Brazil, the NBS program was introduced in stages, and it has been universal (available in all states) since 2013 [30–32]. The Caribbean islands of Guadeloupe and Martinique both have universal NBS for SCD, however, administratively, they are considered territories of France. Jamaica achieved universal status in 2018, but only in the public sector. India, with an estimated 15% of the global SCD births, currently has not implemented nationwide NBS for SCD [33], despite recommendations by government driven programs, and multiple pilot studies showing the feasibility of establishing such programs in rural settings [34–38]. Sub-Saharan Africa accounts for 79% of new SCD cases in the world, and this proportion is projected to increase to 88% by the year 2050 [33]. In this region, while there are a few regional screening programs (Angola, Ghana, Uganda) [39–41], the coverage remains incomplete [42]. Due to global migration and the resulting increasing prevalence of hemoglobinopathies in Europe, some European countries have initiated screening for hemoglobinopathies and G6PD [29, 43]. Of the Western European countries, Spain, France, the Netherlands, Monaco, and the United Kingdom are the only ones with universal coverage.
The screening for SCID was piloted in 2008 in Wisconsin [44], and as of December 2018, it is fully implemented by all US states. Universal screening is also available in Israel, and several pilot programs are ongoing in Europe, the Middle East and Asia [45–48]. In addition to early detection of SCID, implementation of SCID NBS has allowed for an accurate measurement of its incidence. With 1:58,000 births, its incidence has almost doubled from what was previously estimated (1:100,000 births), highlighting that SCID was previously underdiagnosed in the absence of unbiased population screening [49].
Blood Disorders Screened via NBS
In the US, approximately 100,000 people are affected with SCD (~1/360 African American births) [50]. Of the 300,000–400,000 annual new hemoglobinopathy cases worldwide, approximately 80% have SCD and 20% have thalassemia (13% with β-thalassemia and 7% with α-thalassemia) [51]. The prevalence varies by region of the world and it is estimated that 80% of these affected births occur in malaria-endemic LMICs [52]. While sickle variants are predominantly seen in sub-Saharan Africa, thalassemia mutations have high prevalence in the Southeast Asian region. The overlap in the geographical distribution of malaria and hemoglobinopathies is attributed to the malarial-protective effects of both sickle cell trait (HbAS) [53], and α-thalassemia trait [54]. The malarial protection conferred by HbAS appears to be due to increased clearance of infected erythrocyte and reduced parasite growth, rather than decreased infectivity [55]. This protection is, however, lost when the sickle trait mutation is co-inherited with α-thalassemia trait [56].
Sickle Cell Disease
Sickle cell disease (SCD) is an autosomal recessive condition in which an abnormal sickle hemoglobin (HbS) is inherited either as homozygous (HbSS) or as double heterozygous with other Hb variants (e.g., HbSC, HbSD, HbSE, HbSO) or β-thalassemia (HbS/β-thalassemia). SCD is considered a qualitative hemoglobinopathy, as abnormal sickle globin chains are produced in normal numbers, but how they behave in the RBCs leads to the pathophysiology of the disease. Polymerization of HbS in certain conditions (e.g., hypoxia, acidosis, dehydration), results in sickling of RBCs. Sickle erythrocytes have abnormal rheology and are not very pliable, causing vascular obstruction, or microvascular vaso-occlusion. This vaso-occlusion causes both acute and chronic complications. Acute complications from the disease include recurrent episodes of pain, acute chest syndrome, priapism, acute splenic sequestration, and overt stroke. Endothelial dysfunction mediated by hemolysis of sickle RBCs and ischemia-reperfusion injury secondary to microvasculature occlusion, causes ongoing end-organ damage, constituting the chronic complications of the disease [57]. Among the chronic organ damage observed, the brain (cognitive dysfunction, silent cerebral infarcts), spleen (asplenia), kidneys (sickle nephropathy), lungs (obstructive and restrictive disease, pulmonary hypertension), heart (sickle cardiomyopathy), bones (low mineral density, osteonecrosis), and eyes (sickle retinopathy), are particularly affected. The amount of fetal hemoglobin (HbF, α2γ2) present is of prognostic significance and ameliorates disease severity, forming the basis for therapies which can induce its production, such as hydroxyurea [58]. While HbSS and HbS/β° thalassemia genotypes have a severe phenotype, the compound heterozygous states (HbSC, HbSE) may have a less severe course which might correlate with the proportion of sickle versus non-sickle hemoglobin.
Thalassemia
Thalassemia is a quantitative hemoglobinopathy where there is either decreased globin production (α°, α+, β°, β+ deletional mutations) or globin structural instability (αConstant Spring, βLepore non-deletional mutations). Decreased synthesis of α globin chain affects production of stable heterotetramers (HbA = α2β2, HbA2 = α2δ2, HbF = α2γ2) and an excess of unbound γ and β globin chains, which polymerize to form homotetramers β4 (HbH) and γ4 (Hb Bart, also called fast band). These homotetramers have high oxygen affinity resulting in tissue hypoxia. In addition, owing to their decreased stability, they precipitate in erythroid precursors and circulating erythrocytes, resulting in ineffective erythropoiesis and peripheral hemolysis.
The clinically significant α-thalassemias include hydrops fetalis (four-gene deletion, −/−) where fetal/newborn demise from severe anemia occurs if not rescued by intra-uterine or post-delivery transfusions [59]. Deletional HbH occurs when three α genes are deleted (α−/−). This form of HbH tends to have a milder clinical course than the one resulting from homozygous (αTsaudiα/αTsaudiα) or compound heterozygous (αTsaudiα/αAgrinioα), (−/αConstant Springα) non-deletional α mutations. In non-deletional HbH, growth deficits, splenomegaly, and transfusion requirements begin in infancy compared to deletional HbH mutations, where they occur after the first decade of life [60, 61]. Non-clinically significant forms of α-thalassemia include α-thalassemia trait (two-gene deletion, either in cis, αα/− or in trans, α−/α−), or the silent carrier (one-gene deletion, α−/αα). Alpha-thalassemia trait is common in African Americans, who primarily have the trans deletion, as opposed to Southeast Asians who have the α deletion in cis. In β-thalassemia, the α:β globin chain imbalance diminishes production of HbA, causing it to either be absent (thalassemia major: β0β0) or decreased (thalassemia intermedia β0β+, β+β+, and thalassemia trait ββ0, ββ+). Additionally, elevated HbA2 levels from increased δ-globin synthesis is an important parameter for identifying β-thalassemia trait, but may be falsely low in the presence of iron deficiency anemia [62]. Beta-thalassemia major usually becomes symptomatic (severe anemia requiring transfusions) in the first 6 months of life, when the switch from fetal (γ) to adult (β, δ) globin expression occurs [63]. Beta-thalassemia intermedia, hemoglobin Lepore (homozygous and compound heterozygous with β-thalassemia), and dominant β-thalassemia mutations can have a variable phenotype and may occasionally require blood transfusions later in life for worsening anemia [64–66]. The co-inheritance of α-thalassemia silent carrier and trait can act as genetic modifiers of β hemoglobinopathies, mitigating clinical severity [67]. While, co-inheritance of α gene triplication mutations might increase their disease severity by worsening the globin chain imbalance [68].
Glucose-6 Phosphate Dehydrogenase Deficiency
Glucose-6 phosphate dehydrogenase (G6PD) deficiency is an X-linked disorder, where mutations in the G6PD gene (~200 mutations) result in many allelic variants with different levels of erythrocyte enzyme activity and clinical severity. G6PD enzyme is crucial for erythrocyte antioxidant defense, as it catalyzes the first step in the pentose phosphate pathway, the only NADPH-producing pathway in erythrocytes. The NADPH generated maintains a reducing environment in the cell by restoring the level of reduced glutathione. G6PD-deficient erythrocytes have increased susceptibility to oxidative stress from certain drugs, infections, naphthalene balls, and fava beans, resulting in acute intravascular hemolysis. This hemolysis is usually self-limiting, as the hemolyzed older red cells are replaced with younger red cells with higher enzyme activity [69]. The more serious consequences of G6PD deficiency can occur in the neonatal period (particularly in the premature born), where failure to promptly manage the resulting hyperbilirubinemia can cause permanent neurological damage (bilirubin encephalopathy or kernicterus). Concomitant mutation in the uridine-diphosphate-glucuronosyl transferase-I promoter gene (Gilbert’s syndrome) can further increase the risk of developing hyperbilirubinemia [70]. The G6PD A-variant (10%–60% enzyme activity) is commonly seen in individuals of African ancestry and is present in approximately 10% of black males in the United States [71]. Despite being considered less severe than the Mediterranean variant (enzyme activity <10%), G6PD A-variant has the potential to cause hazardous hyperbilirubinemia as highlighted in studies in Nigerian neonates in whom this variant is widely encountered [72, 73].
Immunologic Disorders with Hematologic Abnormalities Screened via NBS
Primary immunodeficiency disorders are a group of more than 300 rare, mostly inherited, diseases resulting from defects of the development and/or function of the adaptive or innate immune system [74]. Most patients are diagnosed within the first year of life due to failure to thrive and/or recurrent and persistent infections such as sinusitis, otitis media, pneumonia, thrush, or skin abscesses. Following diagnosis, supportive treatment with prophylactic antimicrobials and intravenous immunoglobulin infusions is started immediately to prevent occurrence of infections before definitive therapy is performed.
Patients affected with SCID, a form of primary immunodeficiency, present with profound defects in cellular and humoral immunity at 4–6 months of life, coinciding with weaning of protective maternal antibodies. Laboratory studies demonstrate a combination of usually absent or nonfunctional T, B, NK cells, and lack of responses to vaccine antigens [75]. SCID is genetically heterogeneous and in the past, was classified based on the presence or absence of T, B, and NK cells. Currently, mutations in approximately 20 genes controlling maturation of the immune system have been identified in various types of SCID [76]. The list of common gene defects is listed in Table 6.1. Diagnosis can be determined by whole-exome or whole-genome sequencing. Typical patients with SCID have absent or very low number of T cells (<300 autologous T cells/microliter) or T cells of maternal origin and none or very low T-cell function (<10% of lower limit of normal) as measured by response to phytohemagglutinin. The number of B and NK cells can vary in different types of SCID (Table 6.1) [77].
Table 6.1 The most common genetic defects in SCID
SCID is inherited in an autosomal recessive pattern in most patients, and adenosine deaminase (ADA) deficiency is the best-known form of autosomal recessive SCID in which infants lack the ADA enzyme necessary for T-cell survival. However, the most common type of SCID is X-linked, which is caused by mutations in the interleukin 2 receptor gamma gene (IL2RG) gene, encoding the common gamma chain (γc). The γc is shared by multiple cytokine receptors necessary for lymphocyte development and function. Despite a wide variety of genotypes, all patients are susceptible to severe infections, and often develop chronic diarrhea and failure to thrive. Persistent mucocutaneous candidiasis is a usual early finding as well as infections with viral pathogens, such as adenovirus, cytomegalovirus, Epstein–Barr virus, norovirus, respiratory syncytial virus, influenza, and parainfluenza. These, along with some opportunistic infection such as Pneumocystis jirovecii, can frequently be fatal [78]. In countries where NBS is not available, patients succumb to infections following live-attenuated vaccines such as rotavirus, varicella, and Bacillus Calmette–Guerin [79]. Hence, early diagnosis is crucial making this disease a target for NBS programs.
NBS Procedures for Hemoglobinopathies
Screening Methods for Hemoglobinopathies
The majority of the hemoglobinopathy screening programs currently use IEF or HPLC as the primary biochemical screening methods, replacing the dual electrophoresis (cellulose acetate/citrate agar electrophoresis) technique. Although more expensive than electrophoresis, IEF, and HPLC are more suitable for NBS programs as they can provisionally identify and precisely quantify a larger number of Hb variants, require small-volume blood samples and are less labor intensive [14]. However, despite their high sensitivity and specificity, they might be unable to identify variants with similar mobility or retention time, and which co-migrate or co-elute. Hence a two-tiered approach is used by NBS programs wherein the initial test is followed by a complementary secondary technique (dual electrophoresis technique, IEF, HPLC, or DNA based assays). Definitive diagnosis can be obtained with molecular techniques such as Multiplex gap PCR assays (for detecting α gene duplications and α deletional thalassemia mutations), reverse dot blot genotyping (to detect β globin variants and β thalassemia mutations), and DNA sequencing [80–82]. While DNA-based testing is part of the screening protocol in a few states (New York, Texas, Washington, California, Mississippi), its application is limited by high cost, complex instrumentation, and longer time required for analysis.
Utilization of these biochemical screening techniques in low-resource countries (sub-Saharan African countries, India) is difficult, due to the lack of laboratory infrastructure, funding and system of care required for standardized sample testing. Point of care (POC) diagnostics, which are easy to operate and interpret, allow for rapid testing at a low cost. However, depending on the test methodology, they may have limitations. Sickledex (based on decreased solubility of sickle hemoglobin) is unable to distinguish between HbAS and HbSS [83], while Aqueous Multiphase System (based on the high density of sickle RBC) [84], and Paper based SCD assay (paper-based solubility assay) [85], are limited in their ability to differentiate between HbSS and compound heterozygote state such as HbSC. On the other hand, Sickle Scan (lateral flow immunoassay using polyclonal antibodies) [86, 87], and HemoTypeSC (immunoassay using monoclonal antibodies) can identify various variants (HbS, HbC) and distinguish between HbSS and HbSC [88]. The recently introduced HemeChip (micro electrophoresis assay) can identify and even quantitate them [89]. These POC diagnostics are still being investigated and their implementation being evaluated.
Interpretation of NBS Results for Hemoglobinopathies
The expression of β-globin gene cluster (ε, γ, β, δ) is developmentally regulated [90]. HbF is the major hemoglobin for most of intrauterine life and is predominant at birth (60%–95%). The transcriptional switch to predominant adult hemoglobin (HbA = α2β2) occurs soon after birth [63]. While the steepest changes in the Hb fractions occur in the first year of life, it may continue beyond infancy at a slower rate [91]. This decrease might be slower with SCD, with mean HbF levels as high as 20% at 1 year of age [92]. The Hb fraction at birth and later during the adult period determines the hemoglobinopathy diagnosis (Table 6.2).
Table 6.2 Newborn and adult hemoglobin fractionation patterns of sickle and non-sickle hemoglobinopathies
| Diagnosis | Birth | Adulthood | Blood count | Parent genotype | ||
|---|---|---|---|---|---|---|
| Hb fractionation | Hb pattern | Hb fractionation | Hb genotype | |||
| Normal | HbF 80%–90%, HbA 10%–20%,* HbA2 0 | FA | HbA 96%–97%, HbA2 <3.5%, HbF 1%–2% | AA | Normal for age and sex | Not applicable |
| Sickling hemoglobinopathies | ||||||
| Sickle cell trait | HbF 80%–90%, HbA ~6%, HbS ~4%, HbA2 0 | FAS | HbA 55–60%, HbS 35%–40%, HbA2 < 3.5%, HbF 1–2% | AS | Normal for age and sex | At least one parent with one or more βs gene (e.g., HbAS, HbSS) |
| Sickle cell trait with alpha thalassemia trait** | HbF 80–90%, HbA ~7%, HbS ~3%, HbA2 0, Hb Bart 5–10% | FAS + Fast Band | HbA 60%–70%, HbS 20%–30%, HbA2 <3.5%, HbF 1%–2% | AS | Mild anemia (Hb 10–12 g/dL and microcytosis (MCV 70–75fL). Target cells seen on the peripheral blood smear | At least one parent with one or more βs gene (e.g., HbAS, HbSS) and both parents with at least one α gene deletion each (e.g., −α/αα, −−/αα, −α/−α) or one parent with at least two α gene cis- deletion (−−/αα, −−/−α) |
| Sickle cell anemia (homozygous βs) | HbF 80%–90%, HbS 10%–20%, HbA 0, HbA2 0 | FS | HbS 80%–90%, HbF 1%–30%,*** HbA2 <3.5%, HbA 0 | SS | Mild to severe anemia (Hb 6–10 g/dL) and normal MCV (80–90 fL) | Both parents with one or more βs gene (e.g., HbAS, HbSS) |
| Sickle cell anemia (homozygous βs) with alpha thalassemia trait** | HbF 80%–90%, HbS 10%–20%, HbA 0, HbA2 0, Hb Barts 5%–10% | FS + Fast Band | HbS 80%–90%, HbF 5%–10%,*** HbA2 <3.5% (but may be higher), HbA 0 | SS | Mild to moderate anemia (Hb 8–11g/dL) and microcytosis (MCV 70–75 fL). Target cells seen on the peripheral blood smear | Both parents with one or more βs gene each (e.g., HbAS, HbSS) and both parents with at least one α gene deletion each (e.g., −α/αα, −−/αα, −α/−α) or one parent with at least two α gene cis- deletion (−−/αα, −−/−α) |
| Sickle beta zero thalassemia | HbF 80%–90%, HbS 10%–20%, HbA 0, HbA2 0 | FS | HbS 80%–95%, HbF 5%–10%,*** HbA2 4%–6%, HbA 0 | Sβ0 thalassemia | Mild to severe anemia (Hb 7– 9g/dL) and microcytosis (MCV 60–80 fL). Target cells seen on the peripheral blood smear | One parent with one or more βs gene (e.g., HbAS, HbSS) and the other with at least one β0 gene (e.g., β0β0 or βAβ0) |
| Sickle beta plus thalassemia | HbF 80%–90%, HbS ~7%, HbA ~3%, HbA2 0 | FSA | HbS 50–80%, HbA 5%–30% (occasionally higher, but always lower than HbS), HbF 1%–20%,*** HbA2 4%–6% | Sβ+ thalassemia | Moderate anemia (Hb 9–12g/dL) and microcytosis (MCV 60-80 fL). Target cells seen on the peripheral blood smear | One parent with one or more βs gene (e.g., HbAS, HbSS) and the other with at least one β+ gene (e.g., βAβ+, β+β+, or β+β0) |
| Sickle hemoglobin C disease | HbF 80%–90%, HbS ~4.5%, HbC ~4.5%, HbA 0, HbA2 0 | FSC | HbS 45%–50%, HbC 45%–50%, HbF 1%–6%,*** HbA2 <3.5%, HbA 0 | HbSC | Mild anemia (Hb 9–14g/dL) and microcytosis (MCV 70–75). Target cells seen on the peripheral blood smear | One parent with one or more βs gene (e.g., HbAS, HbSS) and the other with one or more βC gene (e.g., βCβC, βAβC) |
| Sickle hemoglobin E disease | HbF 80%–90%, HbS 6%, HbE 4%,**** HbA 0 | FSE | HbS 55%–60% HbE ~30%–35%,**** HbF 1-5%,*** HbA 0 | HbSE | Mild anemia (Hb 8–14 g/dL) and microcytosis (MCV 71–97 fL). Target cells seen on the peripheral blood smear | One parent with one or more βs gene (e.g., HbAS, HbSS) and the other with one or more βE gene (e.g., βEβE, βAβE) |
| Sickle cell with δβ0 thalassemia | HF 80%–90%, HbS 10%–20%, HbA 0, HbA2 0 | FS | HbS 60–80%, HbF 15%–20% ***(heterocellular pattern), HbA2 <3.5, HbA 0 | HbS/δβ0 thalassemia | Mild anemia (Hb 10–12 g/dL) and microcytosis (MCV 76–85 fL) | One parent with one or more βs gene (e.g., HbAS, HbSS) and the other with at least one δβ0 gene |
| Sickle cell with hereditary persistence of fetal hemoglobin (HPFH) | HbF 80%–90%, HbS 10%–20%, HbA 0, HbA2 0 | FS | HbS 60%–70%, HbF 25%–35% ***(pancellular pattern), HbA2 <3.5, HbA 0 (deletion-HPFH) | Hb S/HPFH | Mild or no anemia (Hb 10– 15 g/dL) and microcytosis (MCV 68–88 fL) | One parent with one or more βs gene (e.g., HbAS, HbSS) and the other with homozygous or heterozygous non-deletional HPFH mutation |
| Non-sickling hemoglobinopathies | ||||||
| Beta thalassemia trait (minor) | HbF 80%–90%, HbA 10%–20%, HbA2 0 | FA | HbA 85%–90%, HbA2 5%–7%,**** HbF 2–7% | Hbββ0 or Hbββ+ | Microcytic (MCV 60–80 fL) and mild anemia 12–14 g/dL. Target cells seen on the peripheral blood smear | At least one parent with one or more β+ or β0 gene |
| Beta thalassemia intermedia | HbF 80%–90%, HbA 3%–10%, HbA2 0 | FA | HbA 70%–90%, HbF 10–35%, HbA2 5%–7%**** | Hbβ+β+ or Hbβ0β+ | Moderate to severe anemia (Hb 7–10 g/dL), microcytosis (MCV 50–80 fL) and hypochromia (MCH 16–24 pg). Target cells, basophilic stippling, anisocytosis, poikylocytosis, and polychromasia seen on the peripheral blood smear | Both parents with one or more β+ or β0 gene each |
| Beta thalassemia major (Cooley’s anemia) | HbF 100%, HbA 0, HbA2 0 | F only | β0β0: HbF 90%–95%, HbA2 <3.5%, HbA 0 β0β+: HbF 70%–90%, HbA ~30%, HbA2 >5% | Hbβ0β0 or Hbβ0β+ | Severe anemia (Hb 3–7 g/dL), microcytosis (MCV 50–60 fL) and hypochromia (MCH 12–18 pg). Marked anisocytosis, poikylocytosis, polychromasia, and nucleated red blood cells seen on the peripheral blood smear | One parent has one or more β0 gene, and the other with one or more β+ and/or β0 gene |
| Hemoglobin E with beta zero thalassemia | HbF 80%–90%, HbE ~10%,**** HbA 0 | FE | HbE 30%–70%,***** HbF 10–60%, HbA 0 | HbE/β0thalassemia | Anemia (Hb <8 g/dL) and microcytosis (MCV 60–70 fL) Peripheral blood similar to Cooley’s anemia | One parent with at least one βE gene, and the other with at least one β0 gene |
| Hemoglobin E with beta plus thalassemia | HbF ~90%, HbE ~6%,**** HbA 4% | FEA | HbE 30%–60%,***** HbA 10%–30%, HbF 4–30% | HbE/β+ thalassemia | Anemia (Hb ~ 9.5 g/dL) and microcytosis (MCV 70–80 fL) | One parent with at least one βE gene and the other with at least one β+ gene |
| Alpha thalassemia silent carrier | HbF 80%–90%, HbA 10%–20%, HbA2 0, Hb Bart ~1%–2% | FA | HbA 96%–97%, HbA2<3.5%, HbF 1%–2% | αα/α− | Normal for age and sex | At least one parent with at least one missing α globin gene (e.g., αα/α− or α−/α−) |
| Alpha thalassemia trait ** | HbF 90%, HbA 10%, HbA2 0, Hb Bart 5%–10% | FA + Fast bands | HbA 96%–97%, HbA2 <3.5%, HbF 1–2% | α−/α− or αα/−− | Mild anemia (Hb 11–13 g/dL) and microcytosis (MCV 65–80 fL) | Both parents with at least one missing α globin gene each (e.g., αα/α− or α−/α−) or one parent with at least two α gene cis- deletion (−−/αα, −−/−α) |
| Hemoglobin H disease | HbF 60%–80%, HbA ~5%, HbA2 0, Hb Bart 20%–50% (>25%) | FA + Fast bands | Hb A 80%–90%, HbA2 <3.5, HbF 1%–3%, HbH 1%–40% | α−/−− | Anemia (Hb 7–10 g/dL), microcytosis (MCV 50–65 fL) and hypochromia (MCHC 25–30 g/dL). Basophilic stippling, anisocytosis, poikylocytosis and polychromasia seen on the peripheral blood smear****** | One parent with −−/αα (cis deletion) and the other with at least one missing α globin gene (e.g., αα/α− or α−/α−) |
| Hydrops fetalis | Hb Bart 70%–100%, Hb Portland 10%–15%, HbA 0, HbF 0, HbA2 0 | Fast bands | Not applicable | −−/−− | Can be normocytic with severe hypochromic anemia (Hb 3–8 g/dL) | Both parents have −−/αα (cis deletion) |
Notes: Hemoglobin quantification results as measured by high performance liquid chromatography (HPLC). Hb denotes hemoglobin. βs denotes the sickle mutation in the beta globin gene. βC denotes the C mutation in the beta globin gene. βE denotes the E mutation in the beta globin gene. β0 denotes absence of beta globin gene function. β+ denotes decreased beta globin production.
*HbA is lower is premature babies.
** Alpha thalassemia trait can be a deletion either in cis (−−/αα) or in trans (α−/α−).
*** HbF varies by βs haplotype (e.g., higher in Arab–Indian haplotype) and variants in other locus (e.g., BCL11a) and may be as high as 30% even in the absence of exposure to hydroxyurea.
**** In some cases, HbA2 can be between 3.5% and 5%. It can be even <3.5% in presence of iron deficiency or concomitant δ-gene deletional mutation.
*****HbE co-elutes with HbA2 on HPLC, hence not distinguishable from each other.
****** β precipitate inclusions (“golf-ball inclusions”) can be seen in 35%–90% of red blood cells on HbH preparation.
Limitations of Non-Genetic NBS Testing for Hemoglobinopathies
Although hemoglobinopathy screening methodologies are highly sensitive, they are limited in their ability to detect certain mutations in the β globin gene cluster until the adult Hb pattern has developed. Hereditary persistence of fetal hemoglobin (HPFH) disorder can be identified only beyond the neonatal period, as HbF (>80%) is physiologically high at birth. Additionally, neonatal screening is unable to distinguish between certain mild and severe disease states where the hemoglobin pattern is similar at birth (Table 6.3). Furthermore, the HbA level at birth is highly gestational-age dependent, hence HbA fraction estimation is not a reliable method to distinguish the asymptomatic β thalassemia trait (β0β, β+β) and silent β thalassemia mutations from the symptomatic β thalassemia intermedia (β+β0, β+β+) and dominant β thalassemia (clinically symptomatic heterozygous β-thalassemia mutations) mutations, as they all present with the same hemoglobin pattern on NBS (FA). However, follow-up screening outside of the neonatal period, when the hematological parameters and Hb fractions are expected to have reached adult levels, might help differentiate between these disorders. As the switch from hemoglobin F to A is determined by gestational age, premature infants (gestational age <28 weeks) even without a hemoglobinopathy mutation, have only HbF on their NBS. Additionally, heterozygous sickle cell trait might be confused for a homozygous mutation on the NBS in preterm newborns, especially if the abnormal hemoglobin (HbS or HbC) level is higher than that of HbA [93]. Hence, it is essential to repeat the testing in preterm newborns by age 2 months. Parent testing and DNA-based testing are warranted in some cases. Confirmatory testing with HPLC or IEF during infancy might not be able to differentiate certain thalassemia genotypes (β0β0 vs. HPFH vs δβ0 thalassemia or βEβE vs. βEβ0 thalassemia). Hence a definitive diagnosis for at-risk infants <12 months of age, with a suggestive NBS results can be made by confirmatory testing with DNA-based methods [94]. Alpha locus mutations or deletions resulting in α globin deficiency, is reflected by presence of Hb Bart on the NBS. Although highly sensitive, absence of Hb Bart cannot exclude silent thalassemia carrier (1 α gene deletion). This is specially seen with heterozygous α3.7Kb mutation (affecting the α1 gene), where the Hb Bart level might be too low to be measured reliably. The amount of Hb Bart cannot distinguish between the trans and cis thalassemia trait genotypes.
Table 6.3 Phenotype differences in similar NBS pattern results
| Hemoglobin pattern on the NBS | Mild disease phenotype | Moderate or severe disease phenotype |
|---|---|---|
| FS | HbS/HPFH | HbSS, HbS/β0 thalassemia, HbS/β+ thalassemia* |
| F only | Hbβ0/HPFH, homozygous Hb HPFH | Hb β0β0 thalassemia, Hb β0/δβ thalassemia, homozygous Hb δβ0 thalassemia |
| FE | HbEE | HbE/β0 thalassemia |
| FA + Hb Barts | α thalassemia trait** | HbH*** |
Notes: NBS denotes newborn screening.
* Although rare, HbA fraction might be low or undetectable at birth in certain β thalassemia mutations, causing HbS/β+ thalassemia to be misclassified as HbS/β0 thalassemia.
** 2 α genes deleted or mutated.
*** When Hb H is caused by non-deletional α thalassemia, hemoglobin H is not always detectable.
Technical errors might also result in failing to diagnose an underlying hemoglobinopathy on initial testing. This can occur if there is contamination of sample with maternal blood or transfused donor blood. Testing needs to be done on a pre-transfusion sample, and if not available, repeated 3–4 months post-transfusion. Alternatively, DNA-based methods testing can be done [95]. Delay in transporting and analyzing samples can result in denaturation/precipitation of the unstable Hb variant and, therefore, failure to detect it [96]. Degradation of the extracted dried blood sample can lead to less accurate quantification of Hb Barts, and may result in misdiagnosis [97]. Alternatively, cord blood sample can be used for Hb Barts screening, however these samples are not easily transported and there is a risk of maternal blood contamination.
Follow-Up Strategies for Newly Diagnosed Sickling Hemoglobinopathies
In the US, initial screening for SCD is usually performed on DBS collected 24–72 hours after birth or prior to hospital discharge. Some US states require repeat testing with a second sample collected at 1–2 weeks of age. As the structure of the NBS programs varies by state, there are some differences regarding the follow-up algorithms of presumptive positive/abnormal screening results. In general, for all initial screening results showing presence of HbS fraction, the state department refers all new cases to the PCP or the SCD treatment center. Following referral, a fresh sample is performed for confirmatory testing (~6–8 weeks of age). Neonates with confirmatory results concerning for a clinically significant sickle hemoglobinopathy including FS (HbF >HbS), FSA (Hb F> HbS> HbA), FSC (HbF> HbS = HbC), and FSV (HbF> HbS> HbV, where HbV is referred to as the unknown hemoglobin variant other than S or C) patterns, should have care established with a SCD treatment center and prophylactic penicillin initiated by age 2 months. Repeat testing at 6–12 months of age may be performed if the sickle genotype is still unclear or in the absence of parent studies (Fig. 6.1). The impact of newborn screening in reducing early mortality in SCD was largely due to the integration of newborn screening with comprehensive follow-up care [2, 98]. This integration ensures that the affected newborns have access to ongoing routine health maintenance (routine immunization, parental education, monitoring of growth and development, and disease-modifying therapies) and special healthcare services (stroke risk screening, access to blood bank services, and screening for organ damage) required to improve their quality of life and decrease disease morbidity and mortality [99].
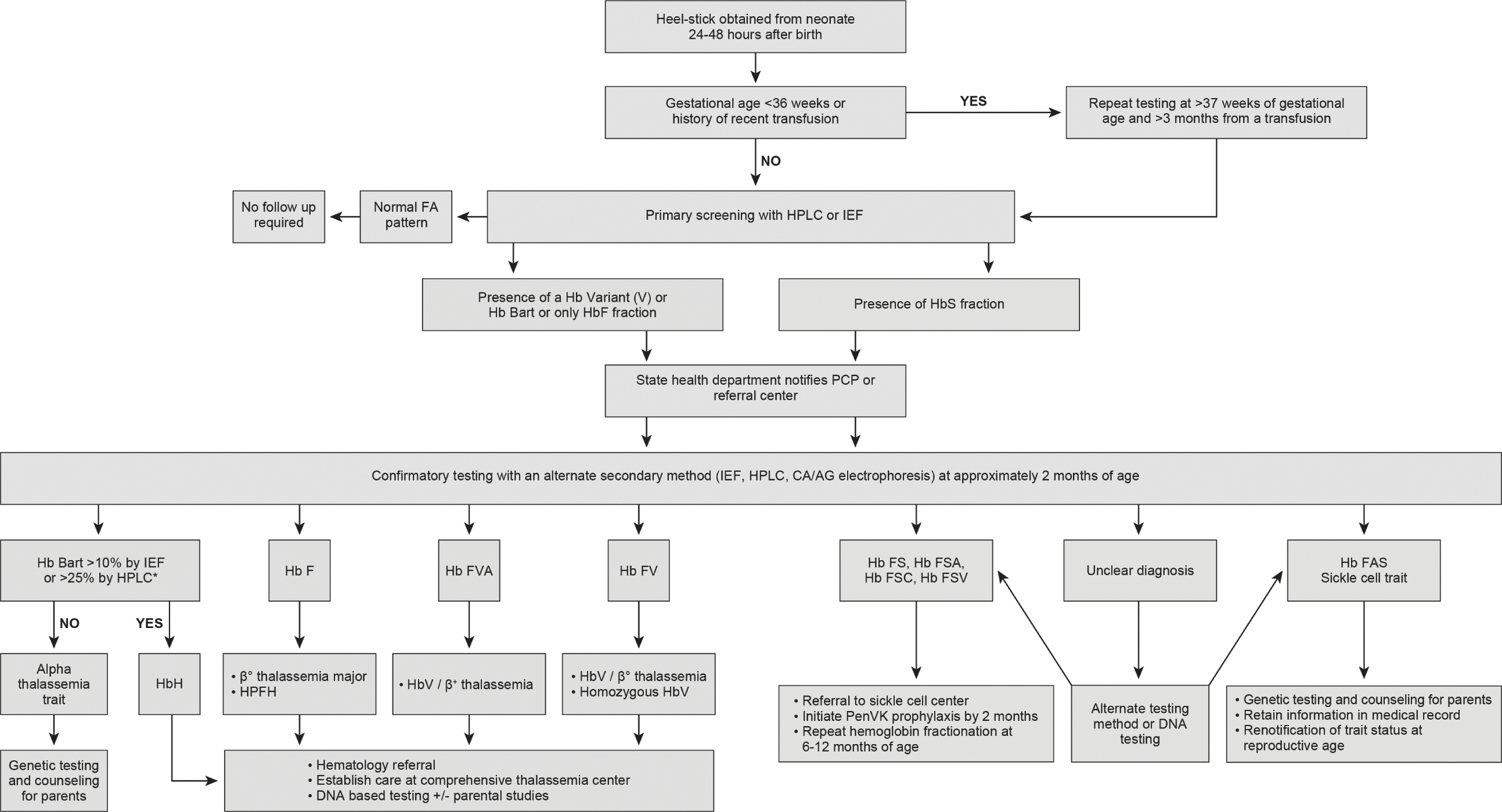
Fig. 6.1 Approach to screening and follow up of hemoglobinopathies in newborns. Flowchart illustrating how to interpret newborn screening hemoglobinopathy results and the follow-up steps to confirm diagnosis and establish early care. HPLC: high performance liquid chromatography, IEF: isoelectric focusing, Hb F: fetal hemoglobin, Hb A: adult hemoglobin A, Hb V: variant hemoglobin, Hb S: sickle hemoglobin, CA/AG electrophoresis: cellulose acetate/agarose gel electrophoresis, PCP: primary care provider, HPFH: hereditary persistence of fetal hemoglobin, *Confirmatory DNA alpha globin gene analysis can also be done to diagnose Hb H disease and alpha thalassemia trait.
Follow-Up Strategy for newly Diagnosed Non-Sickling Hemoglobinopathies
Screening for non-sickle hemoglobinopathies is not mandated by all the NBS state programs in the US. Hence, while incidental findings (Hb variants, Hb Bart, or absence of HbA) suggestive of a clinically significant non-sickle hemoglobinopathy are reported by most states to the PCP and family, the confirmatory testing of these abnormal screening results is directed by the PCP and/or pediatric hematology referral center and not the state. The lack of standard guidelines also has resulted in wide variability in the state based recommendations regarding follow up of abnormal initial screening results (Fig. 6.1). While 7 out of 50 states in the US do not report low levels of Hb Barts, 12 out of 50 states have no follow-up recommendations. The remaining states provide recommendations ranging from complete blood count to alpha globin gene analysis (Wisconsin) [100]. California’s NBS does diagnostic DNA-based second tier testing on samples with clinically significant level of Hb Bart (>25%), for a definitive diagnosis of HbH [101]. Expansion of NBS core panel to include non-sickle hemoglobinopathies would ensure early identification for the clinically significant thalassemia mutations (β0β0, βEβ0, hydrops fetalis, non-deletional HbH) in infancy, and where early intervention (blood transfusion) can improve survival. Integration of this screening with comprehensive follow-up care is also beneficial for the clinically significant thalassemia mutations with varied phenotypic expression where close surveillance, parental education, and long-term follow up ensures decreased disease morbidity and improved quality of life [101]. Early identification of milder mutations (trait and carrier) would help avoid unnecessary iron supplementation for microcytosis in the absence of iron deficiency and aid in identifying couples who are carriers.
NBS Procedures for G6PD Deficiency
The American Academy of Pediatrics (AAP) recognizes G6PD deficiency as a major risk factor for development of hyperbilirubinemia and kernicterus in the newborn, therefore, it recommends pre-discharge bilirubin screening in conjunction with investigation of hyperbilirubinemia risk factors. The AAP recommends that G6PD screening is only done on jaundiced neonates who are at risk of having G6PD based on their family history, ethnicity, and geographic origin or if they are responding poorly to phototherapy [102]. The World Health Organization recommends universal screening for G6PD using fluorescent spot test in areas with a male G6PD prevalence of 3%–5% or more [103]. Reports from working groups have concluded that screening for G6PD using point-of-care biochemical assays during birth hospitalization and timely result notification (<48 hrs) is clinically feasible [104], however, there is no consensus yet regarding how to screen and whether knowledge of G6PD status in the immediate newborn period will prevent neonatal kernicterus [102].
A subset of female neonates who are heterozygous for G6PD-deficient mutation may have a severe presentation with hemolysis, hyperbilirubinemia, and kernicterus [105–107]. In female heterozygotes with random X-inactivation, two erythrocyte populations (~1:1 ratio) are seen, where one population has normal G6PD activity while the other is G6PD deficient and susceptible to hemolysis. Variation in X-inactivation with skewing towards the erythrocyte deficient population may lead to a more severe G6PD-deficient phenotype [108]. Biochemical assays, including the widely recommended semi-quantitative fluorescent spot test, can assess the G6PD enzyme activity by detecting NADPH level [109]. Due to their low cutoff value, they are sensitive in identifying hemizygous males and homozygous females with complete deficiency of G6PD. However, when used to screen female heterozygotes, the result representing the average enzyme activity of both erythrocyte populations (enzyme deficient and sufficient) might be falsely normal, despite the presence of a sizable enzyme-deficient erythrocyte population [110]. Additionally, this assay can be falsely negative when screening G6PD deficient individuals undergoing or recovering from an acute hemolytic event due to the high number of young erythrocytes (with higher G6PD level), necessitating repeat testing in a few weeks [111]. Alternatively, DNA-based analysis can reliably identify female heterozygotes and is also unaffected by a recent hemolytic event. A DNA-based newborn screening assay capable of detecting the five most common G6PD mutations in the USA and estimated to identify approximately 90% of all G6PD-deficient neonates, is currently used by the NBS programs of Pennsylvania and the District of Columbia [112].
DNA technology, though highly sensitive and specific, especially when using DBS elutes, might be limited in their clinical usefulness if the results are not obtained within the typical hyperbilirubinemia risk period [113]. This testing is expensive and can miss new mutations, unless whole exome sequencing is done, and requires technical expertise and infrastructure (a centralized core laboratory) to be scalable. Newer point-of-care screening tools, such as tandem mass spectrometry and digital microfluidics, might help by providing rapid diagnosis at a lower cost and perhaps, enable large scale screening [114, 115].
Related posts:
 Chapter 1 – A Historical Review
Chapter 1 – A Historical Review
 Chapter 7 – A Guide to Identifying the Cause of Anemia in a Neonate
Chapter 8 – Anemia of Prematurity and Indications for Erythropoietin Therapy
Chapter 14 – Fetal and Neonatal Alloimmune Thrombocytopenias
Chapter 7 – A Guide to Identifying the Cause of Anemia in a Neonate
Chapter 8 – Anemia of Prematurity and Indications for Erythropoietin Therapy
Chapter 14 – Fetal and Neonatal Alloimmune Thrombocytopenias
 Chapter 20 – Transfusion Practices
Chapter 20 – Transfusion Practices
 Chapter 21 – Neonatal Leukemia
Chapter 21 – Neonatal Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


