Abstract
Inherited bone marrow failure syndromes (IBMFS) are a rare but important consideration in the differential diagnosis of cytopenias in childhood [1]. However, diagnosis of IBMFS in the newborn period can be challenging because many of the manifestations considered typical for a specific disorder may not yet be present, and in many cases children will not be recognized until later in life. Young children with IBMFS may have one or more cytopenias, congenital anomalies, both, or neither. A high index of suspicion for an IBMFS is required in order to establish the correct diagnosis, determine appropriate clinical management and follow up plans, and provide the family with genetic counseling. Some IBMFS predispose to leukemia or solid tumors; while the development of cancer is uncommon in the newborn period, this risk is an important determinant of subsequent follow up for the child and any affected family members.
Introduction
Inherited bone marrow failure syndromes (IBMFS) are a rare but important consideration in the differential diagnosis of cytopenias in childhood [1]. However, diagnosis of IBMFS in the newborn period can be challenging because many of the manifestations considered typical for a specific disorder may not yet be present, and in many cases children will not be recognized until later in life. Young children with IBMFS may have one or more cytopenias, congenital anomalies, both, or neither. A high index of suspicion for an IBMFS is required in order to establish the correct diagnosis, determine appropriate clinical management and follow up plans, and provide the family with genetic counseling. Some IBMFS predispose to leukemia or solid tumors; while the development of cancer is uncommon in the newborn period, this risk is an important determinant of subsequent follow up for the child and any affected family members.
A comprehensive review of IBMFS is beyond the scope of this chapter; instead, we will focus on disorders most likely to present in the newborn period. We will discuss the clinical findings, diagnostic work up, implications for treatment in the first year of life, and general prognosis. In particular, we will discuss congenital anemia, neutropenia, and thrombocytopenia syndromes, as well as Fanconi anemia (FA), and dyskeratosis congenita (DC), with a brief mention of miscellaneous disorders that may be considered in the differential of bone marrow failure during infancy.
Approach to Evaluation for an IBMFS in the Neonate
The initial approach to evaluation for an IBMFS in the neonate starts with a careful history and physical examination. Additional laboratory and radiographic studies will depend on the index of suspicion. A general algorithm is proposed in Fig. 4.1, acknowledging that a variation to this approach may be appropriate in individual situations.
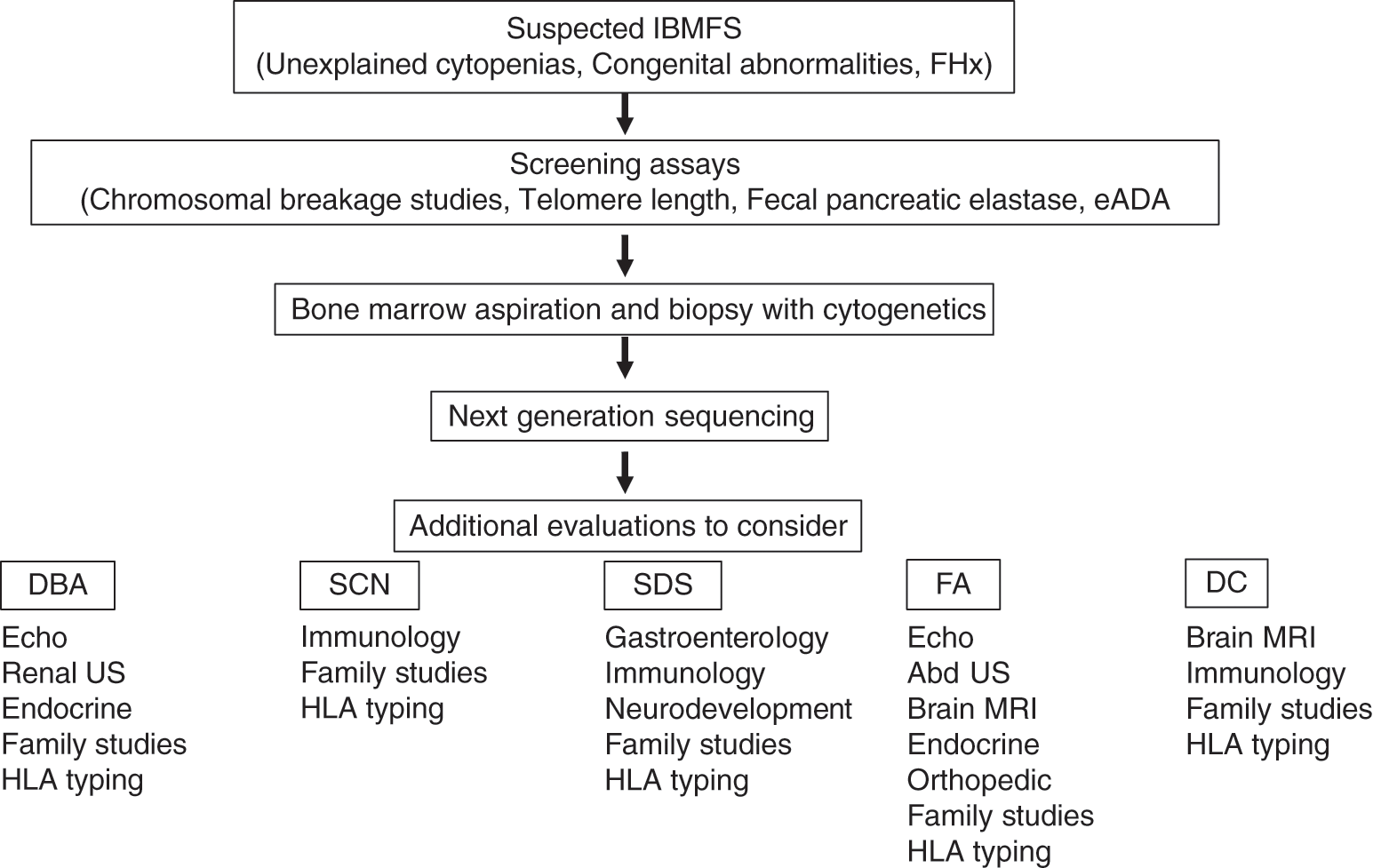
Fig. 4.1 Approach to evaluation of the child with a suspected inherited bone marrow failure syndrome
Physical Examination
Findings on physical exam that may raise suspicion for an IBMFS may include those associated with anemia (pallor, difficulty feeding, tachycardia, in rare cases hydrops), thrombocytopenia (bruising, petechiae, hematoma), or leukopenia (infection of the skin or umbilical cord, recurrent thrush or diaper rash). Alternatively, findings may be limited to physical anomalies such as low birth weight or short stature, dysmorphism, microcephaly, café au lait spots, nail dystrophy, urogenital malformations, or skeletal abnormalities, particularly involving the radial ray and thumbs (Table 4.1). Some findings that are considered to be characteristic of specific diagnoses may not be apparent in the newborn period.
| Frequency of any anomaly | Fanconi anemia | Diamond–Blackfan anemia | Shwachman–Diamond syndrome | Telomere biology disorder | Thrombocytopenia absent radius |
|---|---|---|---|---|---|
| 75% | 40%–50% | 55% | 75% | 100% | |
| Head and face | Microcephaly | Microcephaly | Microcephaly | Microcephaly | |
| Micro-ophthalmia | Hypotelorism | Lacrimal duct stenosis | Micrognathia | ||
| Hypertelorism | Epicanthus | Exudative retinopathy | Hypotelorism | ||
| Ear abnormalities | Micro-ophthalmia | Leukoplakia | Posteriorly rotated low-set ears | ||
| Hearing loss | Broad nasal bridge | ||||
| Micrognathia | |||||
| Cleft lip/palate | |||||
| Central nervous system | Small pituitary | Developmental delay | Cerebellar hypoplasia | ||
| Hydrocephalus | Intracranial calcification | ||||
| Abnormal corpus callosum | Developmental delay | ||||
| Developmental delay | |||||
| Skeletal | Short stature | Short stature | Short stature | Short stature | Short stature |
| Absent or abnormal thumbs | Triphalangeal, bifid or hypoplastic thumbs | Metaphyseal dysplasia of long bones and costochondral junctions | Absent radii | ||
| Absent or hypoplastic radii | Hypoplastic thumbs | ||||
| Congenital hip dislocation | Vertebral anomalies | Ulnar or humeral hypoplasia | |||
| Vertebral anomalies | Narrow thorax | Phocomelia | |||
| Congenital hip dislocation | |||||
| Small patella | |||||
| Bowed legs | |||||
| Cardiac | ASD | ASD | ASD | ASD | |
| VSD | VSD | VSD | VSD | ||
| PDA | Coarctation | PDA | PDA | ||
| Coarctation | TOF | ||||
| Pulmonary stenosis | |||||
| Pulmonary | Tracheoesophageal fistula | Pulmonary fibrosis | |||
| Gastrointestinal | Imperforate anus | Exocrine pancreatic insufficiency | Esophageal stenosis | Cow’s milk intolerance | |
| Esophageal and duodenal atresia | Hepatic fibrosis | ||||
| Fatty pancreatic changes | |||||
| Malrotation | Hepatomegaly | ||||
| Transaminitis | |||||
| Urogenital | Ectopic, horseshoe, absent, or dysplastic kidney | Horseshoe or absent kidney | Horseshoe kidney | ||
| Hypospadias | Duplex ureter | ||||
| Duplex ureters | |||||
| Cryptorchidism | |||||
| Hypospadias | |||||
| Small or absent testes | |||||
| Cryptorchidism | |||||
| Uterine abnormalities | |||||
| Dermatologic | Café au lait spots | Eczema | Lacy pigmentation of the neck and chest | ||
| Hypo or hyperpigmentation | |||||
| Dysplastic nails | |||||
| Sparse hair | |||||
| Endocrine | Growth hormone deficiency | Hypothyroidism | Hypothyroidism | ||
| Hypothyroidism | Abnormal glucose | Osteopenia | |||
| Abnormal glucose | Osteopenia | ||||
| Osteopenia |
Abbreviations: FA (Fanconi anemia), DBA (Diamond–Blackfan anemia), SDS (Shwachman–Diamond syndrome), TBD (telomere biology disorder), TAR (thrombocytopenia absent radius), ASD (atrial septal defect), VSD (ventricular septal defect), PDA (patent ductus arteriosus), TOF (tetralogy of fallot)
Family History
A detailed family history should be obtained with regard to congenital anomalies, unexplained cytopenias, stillbirths, or spontaneous abortions, and unusual infections in siblings or close relatives [2]. A family history of cancer, particularly at a young age, or excessive toxicity from chemotherapy or radiotherapy could suggest an IBMFS. If the information is available, three generations should be included as some associated conditions such as malignancies, may not develop until an older age. Consanguinity within the family increases the likelihood of an autosomal recessive disorder. Some genetic variants may be present at higher frequency in certain ethnic groups; for example, a variant in FANCC is unique to FA patients of Ashkenazi Jewish ancestry, and has a carrier frequency of greater than 1/100 in this population [3].
Laboratory Evaluation
The general laboratory evaluation of a child suspected of having an IBMFS should include a complete blood count with differential, peripheral smear, and reticulocyte count. It is important to be aware of gestation- and age-specific normal ranges for hematopoietic parameters (see Chapter 24 for reference ranges in neonatal hematology). For example, the lower limit of normal for absolute neutrophil count in a >36 week gestation newborn is 2,700/microliter, whereas for a 28–36 week gestation newborn it is 1,000/microliter. An elevation of the red cell mean corpuscular volume (MCV) is suggestive of a marrow failure disorder, but again one must take into account that some degree of macrocytosis is normal in the newborn period. Additional factors that may affect red cell size include reticulocytosis (because reticulocytes are larger than other red cells), trisomy 21 (associated with an elevated MCV), or concomitant thalassemia (causes red cell microcytosis). Recent transfusion will also affect interpretation of the MCV. Bone marrow aspiration and biopsy are generally required in the evaluation of an IBMFS. However, in the absence of clinically significant cytopenias, this study may in some cases reasonably be delayed until an age where sedation can be performed more safely. Important diagnostic findings from the marrow include paucity or abnormal maturation of specific precursors, generalized hypocellularity or cytogenetic changes. More specific diagnostic studies are reviewed in conjunction with individual disorders below.
Additional Studies
Radiographic studies can be useful to document congenital abnormalities not obvious on external exam. In the setting of suspected IBMFS an echocardiogram, abdominal ultrasound, brain MRI, or plain x-rays may be appropriate.
Counseling
Available genetic testing approaches are discussed in Chapter 15. Once an IBMFS is suspected, involvement of a genetic counselor can facilitate pre- and post-test counseling for the family. A genetic counselor can ensure appropriate testing is being performed, educate the family about the implications and limitations of testing, help to interpret results, identify additional family members who should be tested as well as discuss options for planning future pregnancies. Appropriate counseling is particularly important when evaluating families for disorders that carry a significant risk for malignancy. Family members who are found to be affected should be referred for appropriate follow up.
Specific Disorders
Congenital Anemias
Diamond–Blackfan Anemia
The incidence of Diamond–Blackfan anemia (DBA) is 6–7 per million live births with no ethnic or gender predilection. DBA is an important consideration in the evaluation of neonatal anemia, commonly presenting with macrocytic, hypoproliferative anemia in infants less than 1 year of age. Congenital anomalies are frequent, occurring in 40%–50% of patients (Table 4.1), and may include growth failure, microcephaly, facial dysmorphism (hypertelorism, flat nasal bridge, micrognathia), cleft palate, cardiac, and genitourinary malformations, and abnormal thumbs (triphalangeal, bifid, hypoplastic) [4]. Non-classical presentations of DBA, with a later onset of anemia, other cytopenias or even normal blood counts may be more common than is recognized. The phenotype may vary considerably even within the same family [5]. Registry studies have demonstrated a predisposition to cancer in adult patients, including myelodysplasia and solid tumors such as osteosarcoma and colon cancer, and patients with unrecognized DBA may experience unexpected hematologic toxicity with chemotherapy [6, 7].
Pathophysiology
DBA is a ribosomopathy, resulting from a pathogenic variant or deletion of one of several known ribosomal genes, with RPS19 and RPL5 being the most common [8–10]. Inheritance is autosomal dominant, with approximately 55% of cases being sporadic. There are rare cases of the DBA phenotype associated with pathogenic variants of non-ribosomal genes including GATA1, TSR2, ADA2 and EPO [11–13]. There is no clear relationship between phenotype and genotype, although cleft palate may be associated with abnormalities in RPL5.
Diagnostic Evaluation
A diagnosis of classical DBA can be made if all of the following criteria are present: age less than 1 year, macrocytic anemia, reticulocytopenia, and normocellular bone marrow with a paucity of erythroid precursors [14]. While other cytopenias are not considered classical, neutropenia and thrombocytopenia are sometimes present. Additional criteria can be used to make a diagnosis of non-classical DBA if the above clinical criteria are not met. A family history of DBA or of symptoms suspicious for DBA increases the likelihood of this diagnosis; if there is a known familial mutation then testing of the child is straightforward. The finding of congenital abnormalities commonly seen in patients with DBA supports the diagnosis and should be sought by a careful exam as well as an echocardiogram and abdominal ultrasound. Erythrocyte adenosine deaminase (eADA) is elevated in approximately 80% of patients with DBA [15]. Hemoglobin F is frequently elevated in children with DBA, though it is not specific and in children less than 6 months of age elevation of hemoglobin F can be physiologic. Interpretation of eADA and hemoglobin F results will be confounded by red cell transfusion. Genetic studies are an increasingly important diagnostic tool, particularly in the patient who does not meet classical criteria. It is important to note that the features of many of the IBMFS overlap, thus the screening approach should include chromosomal breakage studies, telomere length and fecal pancreatic elastase to rule out an alternative diagnosis (see Fig. 4.1). Unless there is a known familial mutation, the most efficient approach to genetic testing is to use a next-generation sequencing panel with coverage of the known genes associated with DBA, including copy number analysis of deletions and duplications, as well as other IBMFS. With such a panel, testing will identify a pathogenic variant in about 70% of cases [16].
Management
The cornerstone of therapy in infants with DBA involves the use of red cell transfusions as needed in order to maintain the hemoglobin above 9 g/dl [17]. Additional supportive care in the newborn period includes referral to endocrinology and nutrition for children with poor growth, as well as management of associated cardiac or other congenital defects [18]. Although about 80% of children with DBA will have improvement of anemia in response to glucocorticoids, it is recommended to delay institution of this treatment until after 1 year of age, to minimize adverse effects on growth and neurocognitive development [14]. An earlier trial of steroid therapy can be considered if vascular access or other factors are making transfusions difficult. Children who are transfusion-dependent are at risk for iron overload. Chelation is generally required after 10–15 transfusions or when the liver iron content as measured by MRI exceeds 6–7 mg/g dry tissue weight, usually occurring after 1–2 years of age [17, 19]. Ultimately, bone marrow transplantation may be considered for children who are transfusion-dependent and non-responsive to steroids and who have suitable donors. Decisions around the timing of transplant are complicated by the natural history of the disease which can include spontaneous remission. Approximately 20% of patients experience a remission by the age of 25 years, with 72% of those remitting in the first year of life, irrespective of the severity of their phenotype [19]. While incompletely understood, the mechanisms of remission may include somatic revertant mosaicism, whereby spontaneous correction of the pathogenic allele and subsequent clonal expansion of the revertant cell leads to hematologic improvement [20–22]. Other indications for transplant include the development of myelodysplastic syndrome (MDS) which is most commonly a complication of adulthood. Data from the North American Diamond–Blackfan Anemia Registry indicate a median survival of patients with DBA to be 56 years [6].
Additional Congenital Anemias
While DBA is the most common congenital anemia, additional rare anemias that may be present in the newborn period include deficiency of adenosine deaminase 2 (DADA2), Pearson marrow-pancreas syndrome, and congenital dyserythropoietic anemia (CDA).
Deficiency of adenosine deaminase 2 is an autosomal recessive disorder that typically presents as a vasculitis syndrome, with early onset of fevers, rash and stroke, but patients with a phenotype resembling DBA have been reported [23, 24]. Additional hematologic findings may include neutropenia or thrombocytopenia, marrow aplasia, lymphadenopathy, and immunodeficiency [25]. DADA2 can be diagnosed by measurement of ADA2 enzyme activity in plasma or genetic testing. Patients are usually managed by immunology or rheumatology with therapies such as TNF-alpha inhibitors.
Pearson marrow-pancreas syndrome results from single large scale deletions of mitochondrial DNA (mtDNA) and presents during infancy with sideroblastic anemia, exocrine pancreatic dysfunction, and acidosis [26–28]. Though most cases are sporadic, transmission of mutated mtDNA from an affected mother has been described [29]. Anemia is hypoproliferative and macrocytic. Bone marrow findings include vacuolization of erythroid and myeloid precursors and ringed sideroblasts. Muscle weakness, cataract, renal Fanconi syndrome with organic aciduria and cardiac dysfunction may be present, highlighting this as a disorder of multiorgan dysfunction [30]. Treatment is supportive. More than half of patients with Pearson marrow-pancreas syndrome die before 4 years of age due to complications of infection, marrow failure, metabolic acidosis, or liver and renal failure; those who survive may exhibit hematologic improvement and evolution to Kearns-Sayre syndrome [31].
Congenital dyserythropoietic anemia (CDA) is a heterogeneous group of inherited anemias characterized by ineffective erythropoiesis and morphologic abnormalities in bone marrow erythroblasts [32]. Several types of CDA can be distinguished based on the morphologic findings in bone marrow erythroblasts and identification of the causative genes [33]. CDA type I is most likely to be diagnosed in infancy. Infants may present with normocytic or macrocytic anemia, reticulocytopenia, and persistent jaundice; rarely hydrops may develop in utero [34–37]. Review of the peripheral smear may reveal anisocytosis, poikilocytosis, and occasional nucleated red blood cells. In addition to jaundice, manifestations of ineffective erythropoiesis include splenomegaly, frontal bossing, pulmonary hypertension, cholelithiasis, and iron overload [38–40]. Non-hematologic features in CDA-I may be present in 10%–20% of cases and include missing distal phalanges, syndactyly, and absence of nails [41]. Bone marrow morphology is notable for erythroid hyperplasia and a range of dyserythropoietic changes including binucleation, internuclear bridging, and megaloblastic changes [35]. If available, scanning electron microscopy can confirm the presence of nuclear abnormalities in erythroid cells and may show additional widening of nuclear pores, ‘spongy’ heterochromatin, and invagination of the cytoplasm into the nucleus. Genetic testing can confirm the diagnosis and may obviate the need for electron microscopy. Homozygous or compound heterozygous pathogenic variants in CDAN1 or C15ORF41 are identified in approximately 90% of patients [42, 43]. Treatment of CDA-I is generally supportive, including transfusions and management of iron overload which may develop even in the absence of transfusion due to ineffective erythropoiesis [44]. Interferon-alpha may result in stabilization of the hemoglobin and reduction of ineffective erythropoiesis [45–47], and is recommended in the setting of transfusion dependent anemia. While CDA type I is most likely to be recognized in infancy, CDA type II is the most frequent overall. Bone marrow findings in CDA type II include binucleated erythroblasts and, if electron microscopy is available, the appearance of a double membrane in erythroid nuclei. Abnormalities of band 3 expression on erythrocytes may lead to the misdiagnosis of hereditary spherocytosis [48]. CDA type II is caused by homozygous or compound heterozygous variants in the SEC23B gene. Type III CDA is the rarest subtype, with most patients described belonging to a single Swedish kindred and associated with a heterozygous variant in KIF23; bone marrow findings include multinucleated erythroblasts and gigantoblasts. Additional subgroups have been described and include genetic variants in the erythroid transcription factors KLF1 and GATA1 (CDA type IV), or dyserythropietic anemia as part of a broader syndrome (Majeed syndrome, mevalonate kinase deficiency) [49]. Hematopoietic stem cell transplantation has been successfully performed in patients with CDA but the experience is limited [50].
Congenital Neutropenias
Congenital neutropenia syndromes are a heterogeneous group of disorders, both clinically and genetically. They may be categorized according to bone marrow findings, the presence of extrahematopoietic manifestations and the associated risk for leukemia. Now that molecular diagnosis is available, it is more precise to discuss them according to the affected gene, when known (Table 4.2).
| Genes | Location | Inheritance | |
|---|---|---|---|
| Congenital anemias | |||
| Diamond–Blackfan anemia | RPS19 | 19q13.2 | Autosomal dominant |
| RPL5 | 1p22.1 | ||
| RPS26 | 12q13.2 | ||
| RPL11 | 1p36.11 | ||
| RPL35a | 3q29 | ||
| RPS10 | 6p21.31 | ||
| RPS24 | 10q22.3 | ||
| RPS17 | 15q25.2 | ||
| RPL15 | 3p24.2 | ||
| RPS28 | 19p13.2 | ||
| RPS29 | 14q21.3 | ||
| RPS7 | 2p25.3 | ||
| RPS15 | 19p13.3 | ||
| RPS27a | 2p16.1 | ||
| RPS27 | 1q21.3 | ||
| RPL9 | 4p14 | ||
| RPL18 | 19q13.33 | ||
| RPL26 | 17p13.1 | ||
| RPL27 | 17q21.31 | ||
| RPL31 | 2q11.2 | ||
| TSR2 | Xp11.22 | X-linked | |
| GATA1 | Xp11.23 | ||
| EPO | 7q22.1 | Autosomal recessive | |
| ADA2 | 22q11.1 | ||
| Adenosine deaminase deficiency | ADA2 | 22q11.1 | Autosomal recessive |
| Pearson marrow-pancreas syndrome | Multi-gene deletion | mtDNA | Mitochondrial |
| Congenital dyserythropoietic anemia | CDAN1 | 15q15.2 | Autosomal recessive |
| C15ORF41 | 15q14 | ||
| SEC23B | 20p11.23 | ||
| CDAN3 | 15q21 | ||
| KLF1 | 19p13.13 | Autosomal dominant | |
| Congenital neutropenias | |||
| Congenital neutropenia | ELANE | 19p13.3 | Autosomal dominant |
| GFI1 | 1p22.1 | ||
| HAX1 | 1q21.3 | Autosomal recessive | |
| CSF3R | 1p34.3 | ||
| G6PC3 | 17q21.31 | ||
| JAGN1 | 3p25.3 | ||
| WAS | Xp11.23 | X-linked | |
| Familial platelet disorder with associated myeloid malignancy | RUNX1 | 21q22.12 | Autosomal dominant |
| Shwachman–Diamond syndrome | SBDS | 7q11.21 | Autosomal recessive |
| DNAJC21 | 5p13.2 | ||
| EFL1 | 15q25.2 | ||
| SRP54 | 14q13.2 | Autosomal dominant | |
| Cohen syndrome | VPS13B | 8q22.2 | Autosomal recessive |
| Glycogen storage disease 1B | SLC37A4 | 11q23.3 | Autosomal recessive |
| WHIM syndrome | CXCR4 | 2q22.1 | Autosomal dominant |
| Congenital thrombocytopenias | |||
| Congenital amegakaryocytic thrombocytopenia | MPL | 1p34.2 | Autosomal recessive |
| Thrombocytopenia with absent radii | RBM8A | 1q21.1 | Autosomal recessive |
| Radioulnar synostosis amegakaryocytic thrombocytopenia | HOXA11 | 7p15.2 | Autosomal dominant |
| MECOM | 3q26.2 | ||
| Wiscott–Aldrich syndrome | WAS | Xp11.23 | X-linked |
| Fanconi anemia | |||
| FANCA | 16q24.3 | Autosomal recessive | |
| FANCC | 9q22.32 | ||
| FANCG | 9p13.3 | ||
| BRCA2 | 13q13.1 | ||
| FANCD2 | 3p25.3 | ||
| FANCE | 6p21.31 | ||
| BRIP1 | 17q23.2 | ||
| FANCF | 11p14.3 | ||
| FANCI | 15q26.1 | ||
| PALB2 | 16p12.2 | ||
| FANCL | 2p16.1 | ||
| ERCC4 | 16p13.12 | ||
| MAD2L2 | 1p36.22 | ||
| RFWD3 | 16q23.1 | ||
| UBE2T | 1q32.1 | ||
| XRCC2 | 7q36.1 | ||
| RAD51C | 17q22 | ||
| FANCM | 14q21.2 | ||
| SLX4 | 16p13.3 | ||
| RAD51 | 15q15.1 | Autosomal dominant | |
| FANCB | Xp22.2 | X-linked | |
| Dyskeratosis congenita | |||
| DKC1 | Xq28 | X-linked | |
| TINF2 | 14q12 | Autosomal dominant | |
| TERC | 3q26.2 | ||
| RTEL1 | 20q13.33 | Autosomal dominant/autosomal recessive | |
| TERT | 5p15.33 | ||
| PARN | 16p13.12 | ||
| ACD | 16q22.1 | ||
| CTC1 | 17p13.1 | Autosomal recessive | |
| NHP2 | 5q35.3 | ||
| WRAP53 | 17p13.1 | ||
| NOP10 | 15q14 | ||
Severe Congenital Neutropenia
Severe congenital neutropenia (SCN) includes neutropenia syndromes that are associated with a maturational arrest of myelopoeisis. Neutropenia is present at birth, with absolute neutrophil counts less than 500/microliter and often less than 200/microliter. Infections, including pneumonia, cellulitis, skin or liver abscesses and gastrointestinal infections, are common and may be life threatening [51, 52]. Gingivitis and stomatitis are frequently seen by 2 years of age but may not be present in the newborn period. Children with SCN are also at a significantly increased risk to develop myelodysplasia and leukemia (see below) [53].
Pathophysiology
Heterozygosity for a pathogenic variant in the ELANE gene, encoding neutrophil elastase, is present in about 50% of patients with SCN in European and North American registries [54, 55]. Pathogenic variants in ELANE trigger the unfolded protein response in myeloid precursors, resulting in apoptosis of promyelocytes and myelocytes just as they are starting to produce the aberrant protein [56, 57]. It is notable that complete abrogation of ELANE expression is not associated with neutropenia [58]. Variants involving additional genes have been described in patients with the SCN phenotype including HAX1 (Kostmann’s syndrome, autosomal recessive), WAS (Wiskott–Aldrich syndrome, X-linked recessive), as well as others (CSF3R, G6PC3, JAGN1, GFI1) [59]. There are population differences in the frequency of these mutations; for example, data from a Turkish registry found pathogenic variants in HAX1 to be the most common cause of SCN, reflecting a higher rate of consanguinity in that cohort [60].
Diagnostic Evaluation
In addition to neutropenia, a relative monocytosis and eosinophilia may also be present. Bone marrow examination is remarkable for a maturational arrest of myelopoiesis at the level of the promyelocyte or myelocyte. Promyelocytes may exhibit atypical nuclei and vacuolization of the cytoplasm. Overall bone marrow cellularity is either normal or slightly decreased. Because multiple variants may be associated with the phenotype of SCN, in the absence of a known molecular etiology in the family, the most efficient approach to genetic testing is to use a next-generation panel with coverage of all the genes known to be associated with congenital neutropenia.
Management
The availability of granulocyte colony stimulating factor (G-CSF) treatment has led to improved survival for children with SCN, as in the pre-cytokine era more than 40% of reported patients with SCN died from infection before age 2 years [61]. In general, dosing is adjusted to maintain an absolute neutrophil count (ANC) >500–1,000/microliter with the goal of preventing severe infections. However, even if they are better protected from infection, patients with SCN remain at increased risk for leukemic transformation, with a cumulative incidence of death due to MDS or acute myeloid leukemia (AML) in 22% of patients after 15 years of G-CSF therapy in contrast to 10% of death due to infection [53]. The leukemic risk is highest in patients who require doses of G-CSF greater than 8 mcg/kg per day to maintain a protective ANC. Surveillance with annual bone marrow evaluations is recommended to detect morphologic or molecular changes associated with leukemic progression. Bone marrow transplant should be strongly considered for patients who require G-CSF doses greater than 8–10 mcg/kg per day, have significant infections despite G-CSF, have syndromes known to confer a high risk of AML, or who develop somatic mutations associated with leukemic progression (e.g. CSF3R, RUNX1 or development of monosomy 7) [62], on surveillance bone marrow evaluations [52]. Transplant outcomes are better if transplant is performed prior to the development of overt leukemia [63].
Cyclic Neutropenia
Cyclic neutropenia is an enigmatic congenital neutropenia syndrome in which neutrophil counts fluctuate with a regular periodicity [64]. Frequency is estimated at 1 per 106 in the general population. Children usually present prior to 1 year of age with recurrent fevers, mouth ulcers, adenopathy, and bacterial infections, associated with severe neutropenia. Life threatening infections may occur. In between episodes, patients are typically asymptomatic and this can result in a delay in the diagnosis.
Pathophysiology
Intriguingly, the genetic cause of cyclic neutropenia in most cases can be traced to pathogenic variants in ELANE, the same gene that is affected in SCN. Inheritance is autosomal dominant, and a careful history may reveal an affected parent with a history of mouth sores in childhood that abated as they got older, as longitudinal studies have shown that in some cases the amplitude of cycling decreases with age. The reason for cycling remains unclear but the 21 day periodicity is thought to reflect the kinetics of granulopoiesis.
Diagnostic Evaluation
The hallmark of cyclic neutropenia is a pattern of severe neutropenia occurring every 21 days with recovery in between. Blood counts are obtained 2–3 times a week for at least 6 weeks in order to capture 2 nadirs and define the cycle length. At the nadir, an ANC of near 0 typically persists for 3–5 days and is associated with clinical symptoms. Upon recovery, ANC may approach the normal range. Monocytes can be seen to cycle in an opposite pattern; red cell and platelet counts may also fluctuate. A bone marrow evaluation is important to rule out alternative diagnoses.
Management
As in SCN, treatment of symptomatic patients with cyclic neutropenia includes G-CSF to prevent the episodes of oral ulcers and reduce their risk of infection. Relatively low doses of G-CSF are required, with most patients responding to less than 3 mcg/kg per day. Importantly, unlike SCN, patients with cyclic neutropenia are not at significantly increased risk for leukemia and surveillance bone marrow evaluation is not routinely required. Bone marrow transplant is not generally indicated.
Shwachman–Diamond Syndrome
The incidence of Shwachman–Diamond syndrome (SDS) is estimated at 1/76,000 births, with a slight predominance in males [65, 66]. In contrast to SCN, SDS is a multisystem disorder in which extrahematopoietic symptoms such as failure to thrive, exocrine pancreatic insufficiency and skeletal dysplasia may dominate the clinical picture, especially in the newborn period [67] (see Table 4.1). Neutropenia is usually less severe than in SCN, and neutrophil counts may even be intermittently in the normal range. Other cytopenias including anemia (normocytic or macrocytic), thrombocytopenia or even pancytopenia may be seen, and a variety of immunologic abnormalities are also described. Additional findings relevant to the newborn period may include intrauterine growth retardation, immunologic abnormalities, recurrent infections, hepatomegaly with transaminitis, and developmental delay. Significant phenotypic variability may occur, even within the same family. Shwachman–Diamond syndrome carries a risk for later development of MDS or AML, seen in about 30% of patients over time. Those at highest risk for malignant transformation are children who are less than 3 months old at diagnosis who have persistent severe cytopenias (ANC <500/microliter, Hgb <9 g/dl, and/or platelets <100 K/microliter) [51, 59].
Pathophysiology
Shwachman–Diamond syndrome is considered a ribosomopathy, with approximately 90% of patients having pathogenic variants in the SBDS gene on chromosome 7q11 [68]. The most common pathogenic variants are p.Lys62X and Cys84fs, resulting from conversion events between the SBDS gene and its highly homologous pseudogene. The SBDS protein cooperates with elongation factor 1 (EFL1) to catalyze the release of eIF6 from the 60S subunit of the ribosome [69]. Recently, variants in additional genes involved in this pathway, including SRP54, DNAJC21, and EFL1, have been identified in patients who have the SDS phenotype but lack identifiable SBDS mutations [70–72].
Related posts:
 Chapter 1 – A Historical Review
Chapter 1 – A Historical Review
 Chapter 7 – A Guide to Identifying the Cause of Anemia in a Neonate
Chapter 8 – Anemia of Prematurity and Indications for Erythropoietin Therapy
Chapter 14 – Fetal and Neonatal Alloimmune Thrombocytopenias
Chapter 7 – A Guide to Identifying the Cause of Anemia in a Neonate
Chapter 8 – Anemia of Prematurity and Indications for Erythropoietin Therapy
Chapter 14 – Fetal and Neonatal Alloimmune Thrombocytopenias
 Chapter 20 – Transfusion Practices
Chapter 20 – Transfusion Practices
 Chapter 21 – Neonatal Leukemia
Chapter 21 – Neonatal Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


