Abstract
Neonates frequently suffer from life threatening infections. Immaturity of the immune system increases the vulnerability to infection, and the preterm and term neonatal immune system has specific deficiencies relative to that of an older child or adult [1, 2]. During pregnancy, the physical barrier of the placenta and the maternal immune system protect the developing human fetus from infection. However, maternal infections such as rubella, almost eradicated in developed nations through vaccination [3], or the zika virus, an emerging pathogen [4, 5], can ravage the developing embryo and fetus, leading to life-long disabilities. Furthermore, immaturity of natural barrier systems such as skin, bronchial epithelium and the lining of the gastrointestinal tract compound the weaknesses of the immune system of the premature infant [6, 7]. The importance of interactions between the developing immune system, epithelial barriers, and the microbiome to protect the preterm neonate from infection and promote health is increasingly recognized [8, 9]. Ethical and political concerns limit our ability to study the embryological development of the human immune system to the same depth [10, 11].
Introduction
Neonates frequently suffer from life threatening infections. Immaturity of the immune system increases the vulnerability to infection, and the preterm and term neonatal immune system has specific deficiencies relative to that of an older child or adult [1, 2]. During pregnancy, the physical barrier of the placenta and the maternal immune system protect the developing human fetus from infection. However, maternal infections such as rubella, almost eradicated in developed nations through vaccination [3], or the zika virus, an emerging pathogen [4, 5], can ravage the developing embryo and fetus, leading to life-long disabilities. Furthermore, immaturity of natural barrier systems such as skin, bronchial epithelium, and the lining of the gastrointestinal tract compound the weaknesses of the immune system of the premature infant [6, 7]. The importance of interactions between the developing immune system, epithelial barriers, and the microbiome to protect the preterm neonate from infection and promote health is increasingly recognized [8, 9]. Ethical concerns limit our ability to study the embryological development of the human immune system to the same depth [10, 11].
At the earliest stages of human development, the immune system consists of cells and molecules that are nonspecific in their action and do not have the ability to produce amplified responses with repeat exposure to pathogens [12]. The natural, innate immune system functions during the embryonic stage (first 8 weeks post-conception) and consists of macrophages, granulocytes, dendritic, and natural killer cells. The adaptive immune system, consisting of T- and B-cells, begins to function by the second trimester, but is not at full capacity at birth [13, 14]. This chapter reviews how elements of the innate and adaptive immune system develop, how these elements function together as a whole, and relates immune deficiencies that are present in premature and term neonates to development.
The Evolution of Immunity
The ability to recognize self versus non-self as a way to identify and eliminate invading organisms is a basic requirement of survival. Even the simplest single cell organisms such as bacteria have mechanisms such as the CRISPR/Cas system to fend against invading viruses [15]. In more complex organisms such as mammals, the development of epithelial barriers such as skin, mucosal barriers such as gastrointestinal mucosa, and bioactive substances such as pulmonary surfactant, play an integral role in a newborn evading pathogens. However, organisms that penetrate these barriers have the potential to weaken or even kill a host without functional immune effector cells. Furthermore, organisms have evolved the ability to coexist with commensals, bacteria that also can provide protection against potential invading organisms [16].
The time at which various immune cells appear during fetal development mimics the evolutionary order of the appearance of various immune mechanisms. In the late 1800s, Carl van Beer noted that the common features of the simplest to the most complex organisms were present at the earliest stages of fetal development. Development of the blood is no exception. Blood cells have specific developmental stages that are similar to and related to our evolutionary past. For instance, hemoglobin progresses through larval (embryonic and fetal hemoglobin) and adult stages, through mechanisms that evolved in jawed vertebrates [17].
To better understand the embryological development of the human immune system, scientists have studied the evolution of the immune system studying organisms from the most primitive invertebrates to humans. Systems of natural (innate) immunity are present in primitive invertebrates, whereas adaptive mechanisms first appear in the vertebrates, specifically jawless fish [18]. Phagocytic cells first appeared in invertebrate animals and perform many of the functions that macrophages perform in humans. Phagocytic cells in the starfish produce interleukin-1 [19].
Invertebrate phagocytic cells kill bacteria by producing superoxide via an enzymatic reaction. In humans, absence or reduced function of the homologous NADPH oxidase subunit gp91-phox is the cause of X-linked chronic granulomatous disease [20–22]. Aggregates of immune tissue that are directly associated with epithelial tissues such as the skin, gill regions, and gut tissue in echinoderms and tunicates, are similar to the location of lymphatic tissue in mammals [23].
The ability to discriminate self from non-self in multicellular organisms is important for basic nutrition as well as successful reproduction. Mechanisms to discriminate self from non-self are seen in invertebrate animals such as the primitive cnidarian Hydracinia [24]. Furthermore, the colonial tunicate Botryllus schlosseri achieves allo-recognition and rejection through a protein homologous to heat shock protein [25, 26]. The major histocompatibility complex (MHC), required in the human adaptive immune responses, has a common ancestral origin traced to early chordates. In humans, self-recognition is accomplished by the major histocompatibility system MHC. However, the MHC is found in some fishes, amphibians, birds, and mammals [23].
Adaptive immunity first appears in vertebrates. The most primitive lymphocytes, innate lymphoid cells that defend against infection and contribute to wound healing, but do not express rearranged receptors, have been identified in mammals. This group includes natural killer (NK) cells, and cells similar to NK cells that are present in tunicates, the most primitive of chordates [27]. Lymphocytes that rearrange their receptors are thought to have appeared first in the jawless fishes, ancestors to lampreys, and the more specialized T- and B-cells in ancestors of sharks [23]. Both jawed and jawless vertebrates have prototypic T- and B-like lymphocytes which suggest that their origin comes from a common ancestor [28].
Origin of Hematopoietic Stem Cells during Fetal Development in the Mouse
Much of what is known about the biology of the earliest embryological development of the immune system is inferred from the studies in mice. Three waves of hematopoiesis occur in mammals, including humans. The first wave, also described as primitive hematopoiesis, produces primitive erythrocytes, megakaryocytes, and macrophages [29]. Blood cell formation originates in multiple sites in the mouse embryo from “hemogenic endothelium.” The earliest production of blood arises from mesoderm in several sites, including the yolk sac, allantois, vitelline artery and lateral plate [30]. Transcription factors such as SCL/Tal1 [31–33], LMO2/RBTN-2, [34, 35], and AML-1/Runx 1 [30], are critical for the differentiation of hematopoietic cells from primitive mesoderm.
Primitive hematopoiesis results in the production of erythrocytes which are larger than definitive erythrocytes, contain more hemoglobin and are nucleated when they first circulate [36, 37]. These erythrocytes are very well suited for capturing and carrying oxygen in an oxygen poor environment. In addition, primitive megakaryocytes small diploid are produced that are morphologically distinct from adult, bone marrow-derived megakaryoctyes which are polyploid [29].
Macrophages are the sole immunocyte produced in the yolk sac as a result of primitive hematopoiesis [38]. Interestingly, in the mouse, the yolk sac is the common origin of Kupffer cells in the liver, microglia, and dendritic cells, and these persist into adulthood. In contrast, alveolar macrophages in the lungs are progressively replaced by bone marrow-derived macrophages [39]. Recently, a subset of B-cells called innate like atypical B-cells have been described. They have highly restricted immunoglobulin repertoires and reside in epithelial lined cavities such as the peritoneum. The production of these cells, which are critical to neonatal immune responses, occurs from stem cells developmentally restricted to the fetal liver [40, 41].
While able to contribute to the short-term production of primitive erythropoiesis, hematopoietic stem cells (HSCs) from the yolk sac are unable to provide for hematopoiesis long term in the fetal liver or bone marrow, and are unable to reconstitute irradiated mice. The question whether HSCs arise within a given site and colonize other sites or arise independently in each hematopoietic site is controversial. The fact that HSCs from the yolk sac are unable to produce lymphocytes supports the second model; specifically, that the yolk sac and bone marrow independently produce HSCs [42]. However, the ability to track HSCs in mice indicates that definitive HSCs are present in both the yolk sac and intraembryonically in the splanchno pleura/aorta, gonads, and mesonephros (P-Sp/AGM), named for the structures which these areas differentiate into. After generation, these HSCs migrate to and successively colonize the fetal liver, spleen, and bone marrow (Fig. 3.1) [43–47]. The Runx1/AML-1 gene, a proto-oncogene involved with terminal erythroid and megakaryocyte development also plays a crucial role in establishing definitive hematopoiesis [48–50].

Fig. 3.1 Establishment of hematopoietic stem cells. Abbreviations: AGM, aorta gonad mesonephros, HSC, hematopoietic stem cells.
The second wave of hematopoiesis begins with the appearance of “definitive” hematopoietic cells. This term initially referred to the adult form of erythrocytes. The first adult like polyploid megakaryocytes are seeing during this phase [29]. More recently some have defined wave two as hematopoiesis derived from stem cells and multilineage progenitors that can reconstitute neonatal animals but not adult animals following lethal irradiation [51]. Cells destined to be lymphocytes arise during wave two in the yolk sac and are present in the para-aortic splancnopleura, placenta, and vitelline vessels [52–55].
The third wave of blood formation is defined by the emergence of self-renewing stem cells that can provide long-term, high level multilineage engraftment when transplanted into adult mice. Adult repopulating stem cells appear in the aorto/gonado/mesonephros, vitelline and umbilical arteries, placenta and the head of mice prior to engrafting in the fetal liver and bone marrow [30, 56, 57]. Wave 2 and Wave 3 HSCs colonize the fetal liver and bone marrow and are the source of all blood in the adult mouse [51], with the exception of some tissue specific macrophages that derive from wave 1 [39].
Studies focused on gene expression and epigenetic changes in single hematopoietic stem and progenitor cells have suggested that epigenetic changes lead to differences in fate determination in fetal and adult stem cells are derived from interactions that occur between hematopoietic stem cells and surrounding cells during embryological development. In the mouse aorto–glomerulo–mesenephros (AGM) region, studies using single pre-hematopoietic stem cells from the AGM region were cultured with endothelial cells from the AGM. The progeny of these single cells were able to re-populate all the B-cell populations, including B1a cells. Adult stem cells cultured with AGM cells repopulated B-cell populations excluding B1a cells [58]. The changes in stem cells that allow yolk sac derived stem cells to produce certain tissue specific macrophages but not adult stem cells may be related to specific epigenetic changes that occur in hematopoietic stem cells based on cell–cell interactions in the developing embryo [59]. In fact, when fetal liver or cord blood derived stem cells are transplanted into adult hosts, stem cells initially produce immature low ploidy megakaryocytes [60, 61] and immature T-cells [62]. This leads to delayed engraftment and slow immune recovery after cord blood transplant in humans.
Development of the Human Immune Cells and Organs
In the human fetus, hematopoiesis first begins in the yolk sac (weeks 2–5 post-conception), then moves to the liver (weeks 5–24 post-conception), and finally to the bone marrow (week 11 to adult) [63]. The yolk sac phase begins with blood cells developing from mesodermal cells and adjacent to endothelial cells that later become blood vessels. The yolk sac mainly produces large, nucleated erythrocytes with some primitive megakaryocytes and macrophages [64, 65].
The factors that induce changes in sites of hematopoiesis during embryologic development in humans are largely unknown. Five to six weeks post-conception, CD34 positive hematopoietic stem cells appear in the human placenta, and the placenta becomes a major site of erythropoiesis [66, 67]. Simultaneously, macrophages appear in the liver [68]. It is not clear whether these Kupffer cells are derived from yolk sac stem cells as they are in the mouse [39]. Kupffer cells line the sinusoidal spaces and initially account for approximately 70% of the hematopoietic cells in the liver. These macrophages may be involved in remodeling the hepatic structure to make space for hematopoiesis (which will follow). By 6–7 weeks post-conception, erythrocytes outnumber macrophages. The placenta and fetal liver provide an environment that allows for a massive expansion of hematopoiesis that can provide for the needs of the fetus prior to the development of the bony skeleton as the yolk sac recedes in importance. The fetal liver contains precursor cells that produce the full repertoire of immune cells [69, 70]. The common lymphoid progenitors (CLP) generate a full spectrum of lymphoid cells, including T-, B-, and natural killer cells [71–74].
Eight weeks post-conception, the bone marrow space begins to develop. Hematopoiesis begins in the bone marrow between the tenth and eleventh week. (See Fig. 3.2 for hematopoietic stem cell development of mouse and human.) The bone marrow is colonized by HSCs from three separate sources: the yolk sac, P-Sp/AGM, and fetal liver [71–74]. As in the fetal liver, the first hematopoietic cells to appear in the bones in greatest numbers are macrophages, which appear to carve out the marrow space through chondrolysis 8–9 weeks post-conception [71, 74]. By weeks 10–11 neutrophils are the predominant leukocyte in the bone marrow [73, 74].
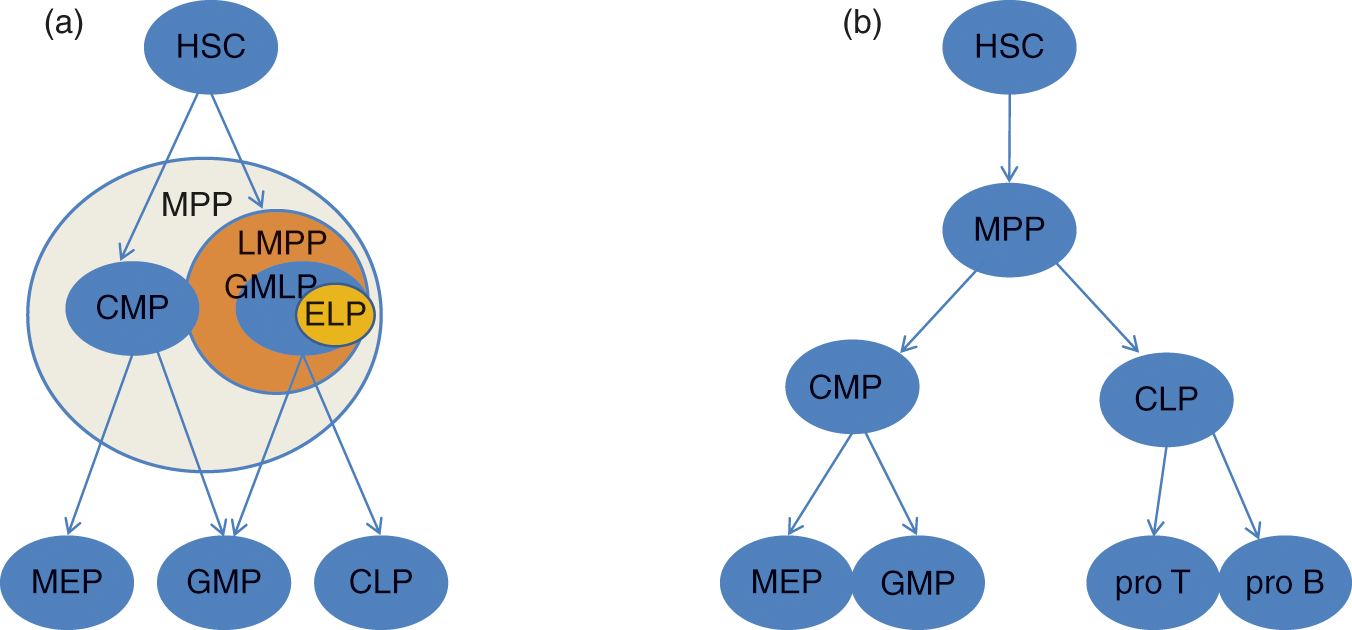
Fig. 3.2 Hematopoietic stem cell development. (a) Mouse hematopoietic hierarchy. (b) Human hematopoietic hierarchy. HSC, hematopoietic stem cell; MPP, multipotent progenitor; LMPP, lymphoid primed multipotent progenitor; CMP, common myeloid progenitor; GMLP, granulocyte monocyte lymphoid progenitor; GMP, granulocyte monocyte progenitor; CLP, common lymphoid progenitor; ELP, early lymphoid progenitor; MEP, megakaryocyte erythrocyte progenitor; pro T, pro T-cell; pro B, pro B-cell.
At the same time hematopoiesis appears in the bone marrow, the thymus begins to develop. Rudiments of the thymus appears 8 weeks post-conception. The function of the thymus is to support the differentiation of T-lymphocytes. Progenitors of the T-cells originating in the fetal liver and later from the bone marrow migrate to the thymus beginning 8–9 weeks post-conception [75, 76]. By the tenth week, lymphocytes constitute 95% of the cells present in the thymus. Granulocyte precursors and macrophages are also present in the developing thymus.
B-cell precursors appear simultaneously in the omentum and fetal liver at 8 weeks post-conception. By 11 weeks, lymphocytes appear in the spleen. By week 22, 70% of splenocytes are lymphocytes [72]. Lymph nodes first appear around 11 weeks post-conception, and lymphocytes appear within the lymph nodes approximately 1 week later. Gut-associated lymphatic tissue also begins to develop 11–12 weeks post-conception. By 19 weeks, structurally mature but immunologically naive Peyer’s patches appear in the intestines [77].
Innate Immunity
As stated earlier, all organs and cell types of the immune system are present by the eleventh week post-conception, but the function and interaction of these cells are poorly understood. In its earliest stages the immune system consists of cells and molecules that are nonspecific in their action. At 8 weeks post-conception, macrophages, granulocytes, and NK cells are the cellular component of the immune system. The noncellular humoral component consists of complement and passively acquired maternal immunoglobulin. Figure 3.3 provides a pictorial summary of the hyporeactive cellular neonatal innate immunity.
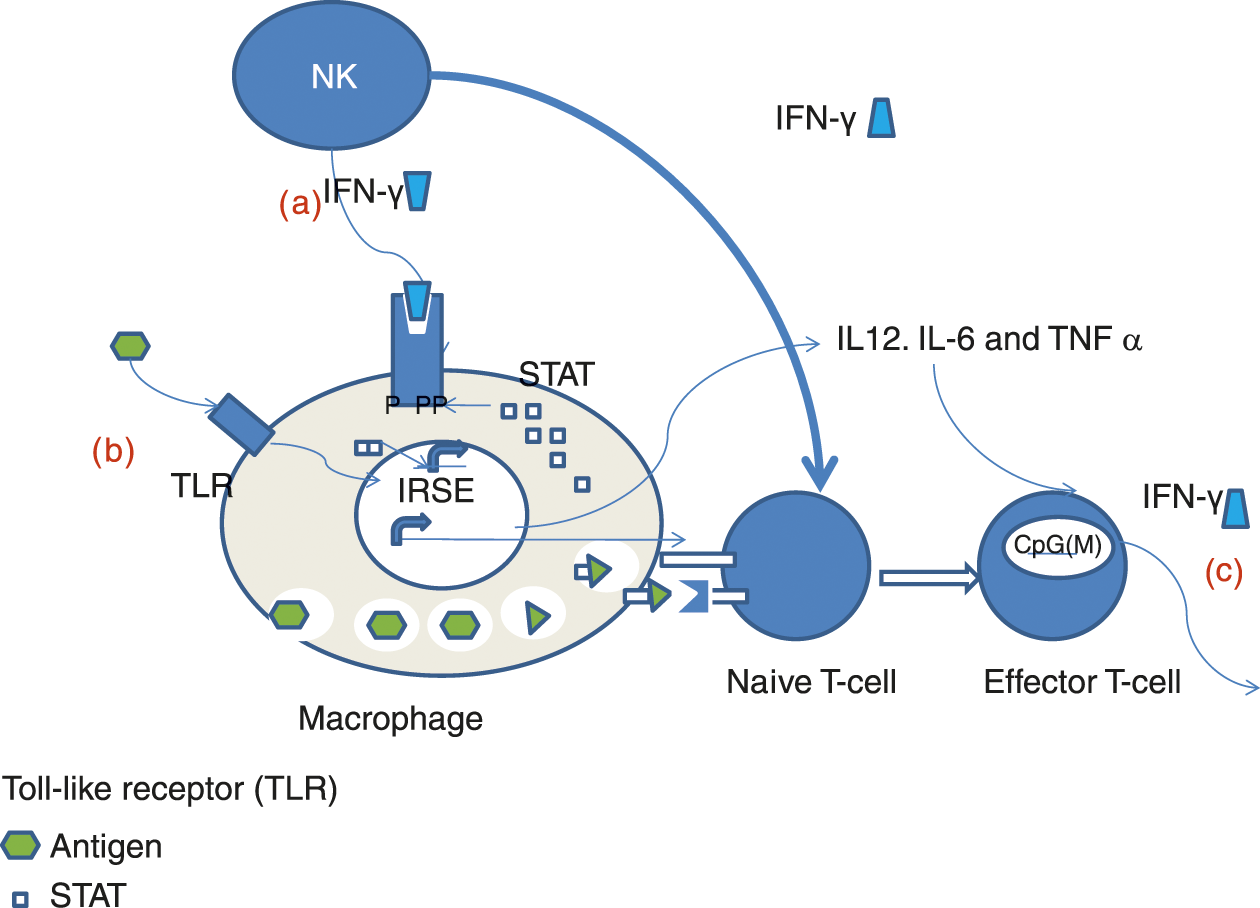
Fig. 3.3 Defects in neonatal innate immune signaling system. (a). Hypoactive response of nenonatal macrophages to activation by IFN– may be related to defective STAT posphorylation (b). Impaired response by neonatal monocytes to multiple TLR ligands (c). Diminished IFN– by neonatal lympocytes may be secondary to hypermethylationof IFN– promoter.
Monocytes/Macrophages
Macrophages play a critical role in the morphogenesis of the fetus. Macrophages are highly mobile, even in their most primitive form. Although a primitive circulatory system is present at 4–5 weeks, there is evidence to suggest that macrophages move through mesenchymal stroma and loose connective tissue. Macrophages shape organs and scavenge debris from apoptotic cells during embryogenesis [78, 79]. They are present in the urogenital ridges where they engulf degenerated cells of the Mullerian duct in the male and the Wolffian duct in the female. They play a similar role in the retina and brain. Fetal hepatic macrophages express Fc receptors and are capable of immune-mediated phagocytosis [80]. In the embryo, Kupffer cells may detoxify circulating endotoxins through their lipopolysaccharide receptor [73].
Macrophages derived from the fetal yolk sac have been shown to constitute the majority of adult tissue-resident macrophages in the liver, brain, lung, and epidermis, demonstrating the importance of these fetal cells over the lifetime of the organism [39, 81]. These fetal-derived cells do not require myb expression for their development in contrast to hematopoietic stem cell-derived macrophages [82]. Lineage tracing experiments showed that fetal yolk sac derived macrophages later give rise to microglia in the brain, while yolk sac monocytes go on to generate the other lineages of tissue-specific macrophages [83].
Studies based mainly on cord-blood derived macrophages demonstrate that neonatal macrophages have diminished immunogenic potential compared to adult cells. Neonatal macrophages have reduced expression of the co-stimulatory molecules CD86 and CD40 leading to poor response to interferon gamma (IFN-γ) and the CD40 ligand [84, 85]. This impaired response leads to an impaired ability for neonatal macrophages to phagocytose pathogens and to activate T cells. Neonatal macrophages also have elevated production of IL-27, which is mainly an immunosuppressive cytokine that blunts the immune response [86]. Neonatal monocytes have been demonstrated to have lower phagocytosis of Escherichia coli when compared to adult monocytes and this is believed to be due to a defect in the toll-like receptor (TLR) activation cascade. In addition, signaling via TLR4 seems to have different requirements in neonates versus adults, where neonates require activity of the TRIF pathway in contrast to adults, who require MyD88 for TLR4 signaling [87]. Neonatal monocytes produce low levels of pro-inflammatory cytokines such as tumor neurosis factor alpha (TNF-α), IL-1 β, or IL-12 in response to lipopolysaccharide (LPS) [88]. Despite the functional immaturity of the macrophage lineage in neonates at birth, the macrophages population in neonates is one of the quickest cellular immune components to mature and approximates adult function within a few weeks [89].
Neutrophils
Neutrophils are also phagocytes. In the adult they internalize and kill microbes. Neutrophils contain granules with reactive oxygen species and hydrolytic enzymes. Neutrophils appear after macrophages in the human fetus. Scattered neutrophil precursors appear in the liver as early as 5 weeks [68, 72, 73]. Neutrophils are also present in small nests in mesenchymal areas prior to onset of hematopoiesis in the bone marrow [72]. Neutrophils first appear in scattered clusters in the bone marrow at the onset of hematopoiesis 10–11 weeks post-conception. By 14 weeks post-conception, neutrophils comprise up to 40% of the hematopoietic cells in the bone marrow [72, 74]. At the time of their appearance in the bone marrow, fetal neutrophils contain myeloperoxidase, an enzyme produced by promyelocytes that is present within the azurophilic granules of neutrophils.
The cycling rates of neonatal neutrophils are elevated compared with adults which suggest an incapacity of precursors to respond to sepsis [90, 91]. Therefore, in the neonate and fetus there is a quantitative defect in neutrophils, discussed in greater detail in Chapter 16. In addition to their quantitative defect, neutrophils have a number of qualitative impairments: transendothelial migration, endothelial adherence, chemotaxis, phagocytosis, intracellular killing, and delayed apoptosis. However, some of these impaired functions may be due more to an increased presence of precursor neutrophils than to impaired function of maturely differentiated neonatal neutrophils [92, 93]. Adhesion and chemotactic defects may be due to the fact that the neonatal neutrophils have lower levels of L-selectin, CD18/CD11b, and CD18/CD11a, which are molecules involved in neutrophil adherence and movement across the vascular endothelium. Chemotactic defects of neonatal neutrophils are caused by a poor response to inflammatory stimuli. Defects in neutrophil phagocytosis are due to reduced levels of serum immunoglobulins. Qualitative neutrophil defects are discussed in Chapter 17.
Natural Killer Cells
Natural killer (NK) cells are phylogenetically primitive lymphocytes that lack specific T-cell receptors, that contain numerous cytoplasmic granules. Natural killer cells develop in secondary lymphoid tissue. Natural killer cells can secrete cytokines and chemokines (IFN-γ, GM-CSF, and TNF-α), and/or kill infected or transformed cells via perforin/granzyme or death receptor. Natural killer cells produce IFN-γ. And IFN-γ triggers the Th1 immune response, activates antigen presenting cells (APC) to upregulate MHC class I expression, activates macrophage killing, and has antiproliferative effects on viral and/or malignant transformed cells. Natural killer cells kill by releasing granules containing esterase and perforin onto the plasma membrane of target cells [94]. They are triggered by the loss of MHC I. Natural killer cells express CD56 on their cell surface but do not express CD3ε, a marker for T-cells.
Natural killer cells appear in the human fetal liver 5 weeks post-conception. As early as 6 weeks, 5%–8% of cells in the fetal liver are NK cells, and by 18 weeks, these increase to 15%–25% of cells. Approximately 10%–15% of cord blood lymphocytes are NK cells. Little is known about the function of NK during the developmental and neonatal period. However, even at birth, NK function is impaired relative to adults [95]. Impaired NK function leads to susceptibility to viral infections [96].
Innate Lymphoid Cells
Innate lymphoid cells (ILC) are lymphoid cells that target conserved pathogen epitopes rather than specific antigens like classic lymphocytes [97]. This lack of recombination-driven antigen recognition makes them ready to respond quickly to infectious insults and classifies ILC as innate even though they are lymphoid in lineage, similar to NK cells. Rather than having directly microbicidal functions like other innate cells including neutrophils and macrophages, ILC function to release cytokines and other bioactive factors that propagate and enhance the immune response. Three categories of ILC have been delineated [98]. The first, ILC1, express the transcription factor T-bet and produce cytokines such as IFNγ that are associated with type 1 immune responses targeting intracellular pathogens [99, 100]. The second, ILC2, generate IL-4 and IL-13 along with other type 2 cytokines that are involved in anti-helminth and allergic responses [101, 102]. The third, ILC3, express RORγt and IL-17A and IL-22 in a type 3 cytokine response which can be involved in extracellular immunity and fibrosis [103–106]. Innate lymphoid cells are active in the fetus and the neonate [107]. And ILC3 in particular are important for gut development and interactions with the microbiota. Innate lymphoid cells also help to prevent homeostatic expansion of T cells in the developing neonatal niche [108].
Complement
The complement system was discovered almost a century ago and is composed of a family of over 25 serum proteins and cell surface receptors that act in an amplifying cascade. The complement system protects against a variety of fungal, bacterial, and viral organisms. The complement system mediates the inflammatory response, serves as a link between the innate and adaptive immunity by increasing B-cell memory, and is important in antibody-dependent killing of micro-organisms. The alternative pathway, which is the more primitive pathway, is initiated without antibodies by endotoxin or other polysaccharides. Complement increases B-cell memory function by: (1) lowering the threshold for activation and maintenance of B-cell survival within the germinal center, (2) causing retention of antigen by the follicular dendritic cell (FDC), and, (3) causing the transport of the immune complex to the FDC by B-cells via complement receptors. There are three general pathways to complement activation: classical, lectin mediated, and the alternative pathway. Typically, the classical pathway requires the binding of the first component of complement (C1) to IgM or IgG antibodies bound to their target antigen. Each pathway activates C3, which causes the terminal cascade to form the membrane attack complex, which lyses invading cells by inserting into the cell membrane.
Various components of the complement cascade appear early in fetal development. C2 and C4 are synthesized in the fetal liver as early as 8 weeks after conception [109]. At 11 weeks post-conception, the receptor for C3B is present on macrophages and neutrophils in the bone marrow, suggesting that neutrophils at this stage have the ability to respond to this chemokine and mediator of inflammation [73]. By 13 weeks, C1, C3, and C5 appear, and by 18 weeks, C7 and C9 are present in the fetal liver. Serum complement levels are low until the third trimester, when they begin to rise and correlate with birth weight and gestational age. At term, complement enzyme levels are approximately 50% of their adult levels [110, 111].
Adaptive Immunity
The last system to mature is the adaptive immune system [89]. These immune components are unique in that they utilize DNA rearrangements to produce unique cell surface receptors that are specific for a certain small amino acid sequence. Other cells that produce major histocompatibility (MHC) molecules use the MHC to present antigenic epitopes to the T and B cells, which causes activation of those T or B cells that are specific to that antigen. The adaptive immune system thus typically takes longer to respond to an infectious insult, because the small number of T or B cells that recognize the antigen then must not only find their cognate antigen but must also clonally expand to generate a cellular immune response. Meanwhile, the B cells in particular can also adapt to the antigen by making small rearrangements to their receptors in a process called somatic hypermutation (SHM), which enhances the avidity of the receptor for the antigen. Figure 3.4 provides a pictorial summary of the neonatal adaptive immune system.
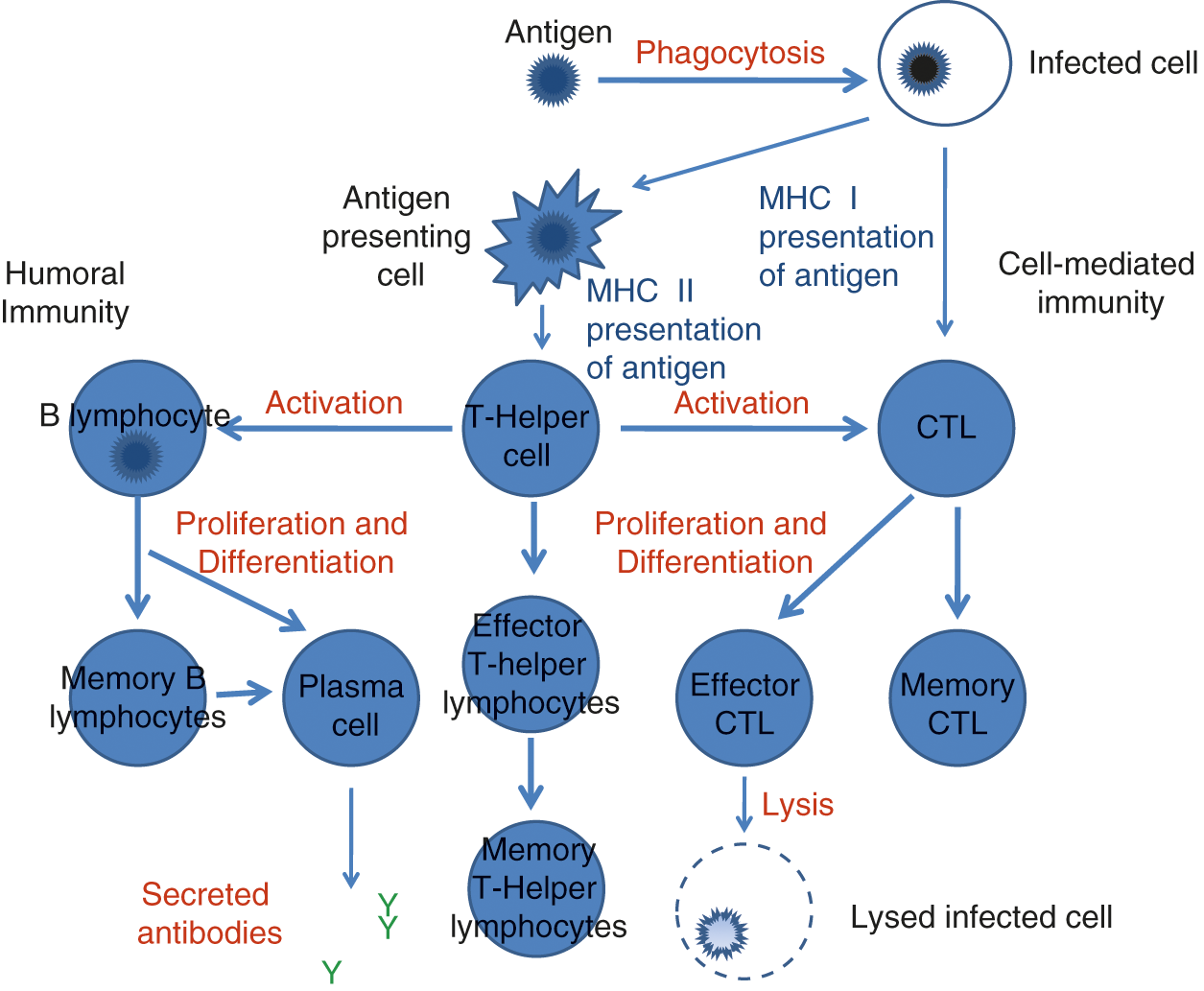
Fig. 3.4 Defects in the neonatal adaptive immune system.
T-lymphocytes
The thymus develops from the endoderm of the third pharyngeal pouch through epithelial mesenchymal signaling and Notch interaction. Lymphoid commitment has been reported to precede thymic development. Haynes described the presence of CD7+ T-cell precursors that are negative for all other T-cell markers in the yolk sac, liver, and thoracic mesenchyme in a fetus 7 weeks post-conception [76]. By 8.5 weeks, cells co-expressing CD7 and CD2, which are progenitors committed to the T-lymphocyte lineage, are present within the thymus. One week later, cells expressing both CD4 and CD8 were present. Progenitor T (pT) cells are found in the circulation and in the fetal liver at 11–12 weeks post-conception, which coincides with their thymic colonization. T-cell maturation depends on the rearrangement and expression of the antigen recognition molecule, the T-cell receptor (TcR). Two types of TcR exist: one composed of a heterodimer called “gamma/delta” and the other called “alpha/beta.” The δ chain is rearranged prior to the β chain [35, 36], and γ/δ T-cells appear prior to the α/β cells during fetal development. However, in the adult the α/β receptor is the most predominant T-cell receptor. The antigen-specific T-cell receptor is complexed to a signal transduction complex called CD3. This complex, when stimulated with a specific antigen, causes the activation and clonal proliferation of T-cells bearing that receptor.
As T-cells mature in the thymus, they progress through three developmental stages: (1) T-cell precursors that do not express CD4 or CD8 antigens; these are called “double negative T-cells,” (2) Cells then become “double positive,” by expressing both CD4 and CD8 antigens, and finally (3) cells then express either CD4 or CD8 and are now mature T-cells [112]. Stage 2 cells exhibit low level surface expression of the TcR–CD3 complex and are positively selected for a rearranged T-cell receptor that recognizes MHC molecules expressed by thymic stromal cells. Stage 3 thymocytes are located in the thymic medulla, with small numbers located within the inner cortex. These cells now express either CD4 or CD8. Cells expressing receptors that recognize “self” MHC or other endogenous antigens presented by the medullary thymic epithelial cells (mTEC) with high affinity undergo apoptosis within the thymus via a process termed “negative selection” [113].
After completing thymic differentiation, mature T-cells enter the bloodstream and migrate to secondary lymphoid organs including the spleen, peripheral lymph nodes, and mucosa-associated lymphoid tissue (MALT). Within the parenchyma of the secondary lymphoid organ, they can be activated after encountering processed antigen. Mature T-cells are able to recognize foreign antigens presented in the context of the MHC. Major histocompatibility class I molecules are expressed by most cells and display mainly intracellular peptides to CD8 positive T-cells. Infected B cells, lymphocytes, macrophages, DC, and other antigen-presenting cells (APCs) express pathogenic antigens in the context of MHC class II molecules. Antigens presented by these cells, which are mainly extracellular in origin, are recognized by the CD4-expressing helper T-cells [114]. Compared with adults, most of the T-cells in the fetus and neonate are CD45RA+, which indicates an immunologically naive state [115]. Fetal T-cells are able to respond to infection, though not at the level found in a newborn or adult. In the congenitally infected fetuses T-cells expressed CD45RO+, a phenotype consistent with previous antigenic activation, as early as 20 weeks after conception [116, 117]. They also expressed the IL-2 receptor-α, another marker of activation. T-cells express CD40 ligand, which is necessary for B-cell activation and class-switching during the second trimester. Durandy found CD40-ligand expression in fetuses from 19 to 28 weeks, but not from 31 weeks to 10 days after birth. CD40-ligand was again detectable 3 weeks after birth [118].
Peripheral blood lymphocytes are capable of cell-mediated lympholysis from 18 weeks post-conception [119, 120]. CD4 to CD8 ratios and alpha–beta to gamma–delta T-cell ratios in cord blood of fetuses from 18 to 34 weeks were similar to newborn and adult levels, suggesting that the altered immune responses are due to intrinsic immaturity in the signaling system of the T-cells [117]. Neonates have higher levels of regulatory T-cells [121], which express CD25 and the transcription factor Foxp3 and are crucial in preventing autoimmunity. Regulatory T-cells (Tregs) may impair the ability of the neonatal immune system to control infections, in particular through inhibition of the activity of antigen presenting cells, however, there is additional evidence that neonatal Tregs may be less functionally competent than adult Tregs [122, 123].
Neonatal susceptibility to many types of infections including enterovirus, Herpes simplex virus, Candida albicans, and Toxoplasma gondii are due in part to impairment of T-cell function. Immaturity of other elements of the immune system and reduced cytokine production contribute to T-cell dysfunction in human neonates. Neonatal antigen presenting cells such as dendritic cells (DC) have been shown to have decreased function compared to adult DC [124]. T cell-derived cytokines control the immune responses in the neonate. Compared with the adult, they have decreased production of certain lymphokines, such as interferon-gamma, IL-4, and IL-5 [125].
Related posts:
 Chapter 1 – A Historical Review
Chapter 1 – A Historical Review
 Chapter 7 – A Guide to Identifying the Cause of Anemia in a Neonate
Chapter 8 – Anemia of Prematurity and Indications for Erythropoietin Therapy
Chapter 14 – Fetal and Neonatal Alloimmune Thrombocytopenias
Chapter 7 – A Guide to Identifying the Cause of Anemia in a Neonate
Chapter 8 – Anemia of Prematurity and Indications for Erythropoietin Therapy
Chapter 14 – Fetal and Neonatal Alloimmune Thrombocytopenias
 Chapter 20 – Transfusion Practices
Chapter 20 – Transfusion Practices
 Chapter 21 – Neonatal Leukemia
Chapter 21 – Neonatal Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


