Abstract
In this chapter, we will focus exclusively on acquired thrombocytopenias that present in the neonatal period. We will discuss the mechanisms underlying some of the most common varieties of neonatal thrombocytopenia, and how the biological differences between neonatal and adult megakaryocytes might contribute to the susceptibility of neonates to develop thrombocytopenia. We will then review the presentation, pathophysiology, and management of the most common etiologies of neonatal early- and late-onset thrombocytopenia, including autoimmune neonatal thrombocytopenia. Alloimmune and congenital causes of neonatal thrombocytopenia will be discussed in detail in Chapters 14 and 15, respectively.
Introduction
In this chapter, we will focus exclusively on acquired thrombocytopenias that present in the neonatal period. We will discuss the mechanisms underlying some of the most common varieties of neonatal thrombocytopenia, and how the biological differences between neonatal and adult megakaryocytes might contribute to the susceptibility of neonates to develop thrombocytopenia. We will then review the presentation, pathophysiology, and management of the most common etiologies of neonatal early- and late-onset thrombocytopenia, including autoimmune neonatal thrombocytopenia. Alloimmune and congenital causes of neonatal thrombocytopenia will be discussed in detail in Chapters 14 and 15, respectively.
Neonatal Platelet Production
In this section, we will review the most important differences between neonatal and adult platelet production (Table 13.1) and will discuss how these differences might predispose neonates to develop thrombocytopenia during illness. The process of platelet production in neonates, as in adults, can be schematically represented as consisting of four main steps (Fig. 13.1):
1. The production of thrombopoietin (Tpo), the most potent stimulator of platelet production, in the liver.
2. The proliferation of megakaryocyte (MK) progenitors (the cells that multiply and give rise to MKs).
3. The maturation of MKs.
4. The release of new platelets by mature MKs.
| Adults | Neonates | |
|---|---|---|
| Tpo concentrations | Very high in hyporegenerative thrombocytopenia | Not as high in SGA neonates as in thrombocytopenic adults |
| Megakaryocyte progenitors | Sparse in the blood | Abundant in the blood |
| Give rise to small colonies | Less sensitive to Tpo | |
| Give rise to large colonies | More sensitive to Tpo | |
| Megakaryocytes (MKs) | Large | Small |
| High ploidy levels | Low ploidy levels | |
| Make more platelets per MK | Make less platelets per MK | |
| MK response to Tpo | Increased MK size/ploidy | No increase in MK size/ploidy |
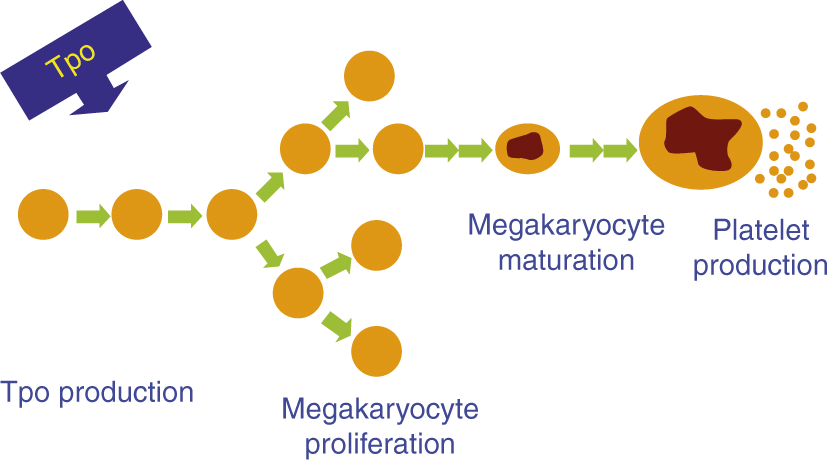
While in essence these steps are the same at all developmental stages, there are substantial differences between neonatal and adult MK biology. Specifically, plasma Tpo concentrations are higher in healthy neonates than in healthy adults [1–3]. Neonatal MK progenitors have a higher proliferative potential than adult progenitors, and generate more MKs per colony [4, 5] and approximately 10 times more MKs per progenitor cell than adult progenitors in vitro [6, 7]. However, neonatal MKs are significantly smaller and of lower ploidy than adult MKs [8, 9], and generate fewer platelets per cell [10]. Importantly, despite their small size and low ploidy, neonatal MKs have been shown to be cytoplasmically mature and contain all of the elements needed for platelet production [6].
Studies in thrombocytopenic adult patients and in animal models have shown that, under normal conditions, the adult bone marrow (BM) responds to increased platelet demand by first increasing the MK size and ploidy and then the MK number [11, 12]. These changes ultimately lead to a two- to eight-fold increase in MK mass. To determine whether thrombocytopenic neonates can mount a similar response when facing increased platelet consumption due to illness, our group evaluated the MK concentration and size in BM samples from thrombocytopenic and non-thrombocytopenic neonates and adults. In this study, thrombocytopenic neonates (unlike thrombocytopenic adults) did not increase the size of their MKs [13]. Similar results were obtained in a study using a murine model of fetal immune thrombocytopenia [14]. In this study, adult mice treated with an anti-platelet antibody (MWReg30) increased the number and size of their MKs as expected, while newborn mice exposed in utero to the same antibody did not increase their MK size. Consistent with these observations, a recent study showed significant differences in the response of newborn and adult mice to a single dose of the Tpo mimetic romiplostim [15]. While adult mice increased the number and size of MKs in the BM and spleen, newborn mice did not increase their MK size, which resulted in an attenuated platelet increment [15]. Taken together, these observations support the notion that MKs in neonates have a limited ability to increase their size in response to increased platelet demand or to Tpo stimulation, which decreases their ability to upregulate platelet production and likely contributes to the predisposition of sick neonates to develop thrombocytopenia. Furthermore, the evidence suggests that platelet production in neonates is dependent on the proliferative potential of their MK progenitors, thus making them highly susceptible to factors that affect MK proliferation.
Neonatal Platelet Function
Multiple in-vitro studies evaluating platelet function in cord blood samples have shown that neonatal platelets are hyporesponsive to most platelet agonists, compared with platelets from adults [16, 17]. However, whole blood assays of hemostasis (such as the bleeding time, the Platelet Function Analyzer, PFA-100®, or the thromboelastogram) revealed that term neonates have normal to slightly enhanced hemostasis compared to adults [18, 19]. While the mechanisms underlying these apparently contradictory findings are not completely understood, it is currently accepted that the higher hematocrits, higher mean corpuscular volumes (MCVs), higher von Willebrand factor (vWF) levels, and presence of ultra large vWF polymers in neonates account for the apparent disparities between the results of platelet function assays and tests of primary hemostasis [20, 21]. Thus, it seems that the platelet hyporeactivity of full term infants should be viewed as part of a delicately balanced neonatal hemostatic system, rather than as a developmental deficiency.
Most of the studies mentioned above only included full term infants, and only a few evaluated preterm neonates at highest risk of bleeding. Bednarek et al. serially evaluated platelet function by flow cytometry in extremely low birth weight (ELBW) infants over the first 2 weeks of life [22]. These investigators showed that platelet responses to agonists and platelet pro-coagulant activity were significantly decreased in ELBW infants during the first days of life compared to adults, but improved significantly by day of life 10–14, to levels only slightly below those of adults [22]. Consistently, del Vecchio and collaborators found that bleeding times performed on day of life one were twice as high in infants born at 24–33 weeks’ gestation compared to infants born at 38–41 weeks’ gestation, but improved by day of life 10 [23]. These observations suggest that preterm neonates may have transient developmental deficiencies in primary hemostasis, but it remains unclear whether these contribute to the pathophysiology of intraventricular hemorrhages.
Specific Neonatal Diseases Causing Thrombocytopenia
Placental Insufficiency and Chronic Intrauterine Hypoxia
Chronic intrauterine hypoxia is the most frequent cause of early-onset thrombocytopenia (within the first 72 hours of life) in preterm neonates. Chronic intrauterine hypoxia is commonly seen in maternal conditions associated with placental insufficiency, such as pregnancy-induced hypertension and diabetes, and manifests in the fetus as intrauterine growth restriction resulting in small for gestational age (SGA) neonates and hematological abnormalities (i.e., thrombocytopenia, neutropenia, polycythemia). The thrombocytopenia associated with this condition is almost always mild to moderate, with platelet counts between 50 and 100 × 109/L. In a large study of SGA neonates with thrombocytopenia presumably due to placental insufficiency and chronic intrauterine hypoxia (i.e., without other identifiable cause), which the authors termed “thrombocytopenia of SGA,” the mean nadir platelet count was 93 × 109/L [24]. In the same study, severely SGA neonates (birth weight <1st percentile for age) had lower platelet counts than less growth restricted infants (Fig. 13.2), and the nucleated red cell count at birth correlated with the severity of the thrombocytopenia. Severe thrombocytopenia secondary to SGA is rare, however, and for that reason other causes of severe early onset thrombocytopenia (such as immune-mediated, infections, or genetic disorders) should be considered if the platelet count is <50 × 109/L. The thrombocytopenia associated with chronic intrauterine hypoxia and intrauterine growth restriction also follows a well-characterized natural course, reaching its nadir by day of life 4, and typically resolving by days of life 10–14. Thus, persistence of the thrombocytopenia beyond this period should also trigger an evaluation for other causes.
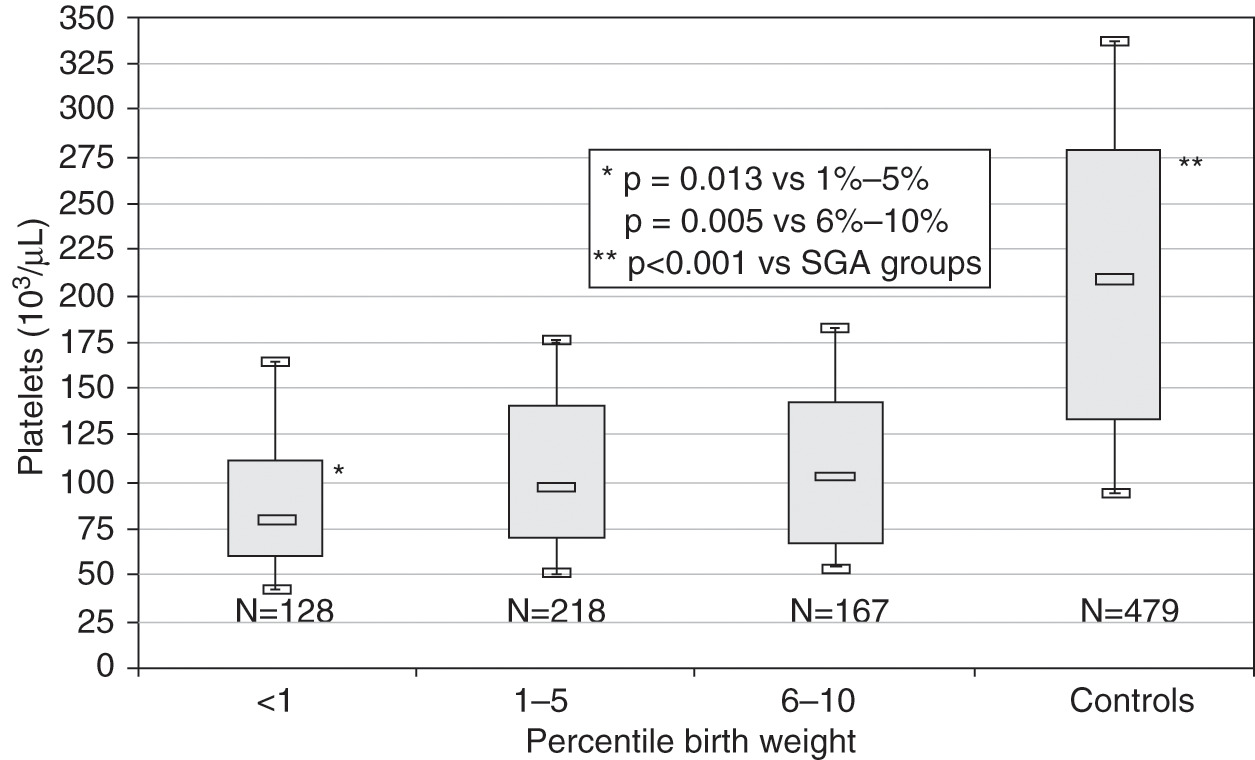
Fig. 13.2 Platelet counts measured on day of life 4 (the mean nadir of platelet count) in SGA neonates according to birth weight percentile. Counts are grouped as neonates <1st percentile, 1st to 5th percentile, 6th to 10th percentile, and 11th to 99th percentile for gestational age (“controls”). In each weight category, the median platelet count is shown, along with the first and third interquartile range (box), and the 10th percentile and 90th percentile values (whiskers).
The pathophysiology of thrombocytopenia in fetuses and neonates exposed to chronic intrauterine hypoxia is not completely understood, but studies have implicated decreased platelet production. Murray and Roberts observed that preterm neonates with early-onset thrombocytopenia had decreased concentrations of circulating MK progenitors compared to their non-thrombocytopenic counterparts [1, 25]. Similarly, Sola and collaborators reported decreased numbers of MKs in the BM of three thrombocytopenic preterm neonates born from pregnancies complicated by placental insufficiency [2]. Contrary to what would be expected, both groups of investigators reported that plasma Tpo concentrations in these neonates were normal to minimally elevated, suggesting that an inadequate up-regulation of Tpo production could contribute to the thrombocytopenia.
The effects of chronic hypoxia on thrombopoiesis in vivo have also been studied in animal models. These studies confirmed the association between chronic hypoxia and thrombocytopenia, and showed reduced MK differentiation of hematopoietic precursors resulting in decreased MK numbers in both adult and newborn animals exposed to hypoxia [26, 27]. Subsequent in vitro studies demonstrated that MK progenitors are not directly damaged by hypoxia, but rather the effects seem to be mediated by non-progenitor cells [28]. Consistently, hypoxia was shown to alter the effects of cytokines on MK progenitors without damaging the MK cells directly [29]. Taken together, these findings suggest that chronic intrauterine hypoxia alters the MK response to cytokines leading to decreased MK production, which improves once the infant is exposed to the normoxic extrauterine environment. This explains the spontaneous resolution of this thrombocytopenia starting on day of life 4–5. Typically, no intervention is needed for the thrombocytopenia in affected infants, but it is important to document the normalization of the platelet count to rule out other possible causes.
Autoimmune Neonatal Thrombocytopenia (Secondary to Maternal ITP)
Early onset thrombocytopenia can also be caused by the transplacental passage of anti-platelet antibodies from the mother to the fetus. Most commonly, these are alloantibodies that react against the fetal platelets, but not the maternal platelets, so that the mother is not thrombocytopenic herself (fetal/neonatal alloimmune thrombocytopenia). The pathophysiology, evaluation and management of this variety of thrombocytopenia are reviewed in detail in Chapter 14.
Other times the neonatal thrombocytopenia is secondary to the passage of antibodies from a mother with immune thrombocytopenic purpura (ITP), or other autoimmune condition associated with anti-platelet antibodies. Several large observational studies published in the 1990s concluded that infants born to mothers with ITP are at low risk of developing severe thrombocytopenia. In one of the earliest studies, Burrows and Kelton found a platelet count <50 × 109/L in 10% of infants born to a mother with ITP, and a platelet count <25 × 109/L in 4.2% [30]. In that study, encompassing 288 infants, no infant suffered an intracranial hemorrhage (ICH) [30]. Other studies reported similarly low incidences of severe thrombocytopenia (defined as a platelet count <50 × 109/L) in neonates born to mothers with ITP, ranging from 8.9% to 14.7%, with ICH occurring in 0% to 1.5% of thrombocytopenic infants [31–33]. In a recent national cohort study conducted in the UK including 107 pregnancies with ITP, no neonates required treatment for thrombocytopenia and there were no cases of neonatal intracranial bleeding. However, the mothers with ITP were at increased risk of severe post-partum hemorrhage compared to the general population [34].
Several studies have tried to identify antenatal predictors of severe thrombocytopenia in the offspring of mothers with ITP. Unfortunately, the fetal or neonatal platelet count cannot be reliably predicted by maternal platelet count, platelet antibody levels, or history of maternal splenectomy. Neonates born to a mother with history of delivering an infant with thrombocytopenia are usually as affected as the first. Attempts to measure the fetal platelet count prior to delivery carry risk and are not recommended. There is also no evidence that cesarean section is safer for the fetus than uncomplicated vaginal delivery (although forceps or vacuum assist deliveries should be avoided), and thus the current recommendation is to treat the maternal ITP appropriately, but to use only obstetrical indications for cesarean delivery. Interestingly, a recent study reported the use of a novel recombinant human thrombopoietin (rTPO) to manage ITP during pregnancy. In this study of 31 patients, 74% responded. Furthermore, rTPO was well tolerated and no problems were observed in the infants born to treated mothers [35]. This paved the way for the use of a new potential therapy to manage ITP during pregnancy.
While only a small percentage of neonates are born with severe thrombocytopenia in the setting of maternal ITP, mild to moderate thrombocytopenia is more common. Importantly, the platelet count typically decreases after birth, reaching a nadir between days 2 and 5 [36]. For that reason, the recommendation is to obtain a platelet count immediately after delivery (from the cord or the baby), and to follow platelet counts in all infants with mild or moderate thrombocytopenia. A head ultrasound should be obtained on all infants with a platelet count <50 × 109/L at birth. Recommendations from the International Consensus Report on Management of ITP are to treat neonates with either clinical bleeding or a platelet count <20 × 109/L with a single dose of intravenous immunoglobulin (IVIG, 1 g/kg), which can be repeated if necessary [32]. Major bleeding should be treated with platelet transfusions in addition to IVIG. Severe thrombocytopenia and major hemorrhage secondary to maternal ITP are rare, so evaluation for alloimmune thrombocytopenia or other causes of thrombocytopenia should be considered in those cases [32].
Neonatal thrombocytopenia secondary to maternal ITP can last for months and requires long-term monitoring and occasionally a second dose of IVIG at 4–6 weeks after birth. Interestingly, a recent study suggested that antiplatelet antibodies from ITP mothers are transferred to the fetus by breastmilk, and are associated with neonatal thrombocytopenia persisting more than 4 months, which disappears following discontinuation of breastfeeding [37].
Asphyxia
Several studies have established an association between perinatal asphyxia and thrombocytopenia [38, 39]. The thrombocytopenia is typically self-limited and mild to moderate, with platelet counts between 50 and 100 × 109/L that improve spontaneously and normalize by days 19 to 21 [40]. In a cohort of 171 neonates with perinatal asphyxia, Boutaybi et al. found early-onset thrombocytopenia (within the first 48 hours of life) in 51% [41]. Multiple logistic regression analysis identified a significant independent association between early-onset thrombocytopenia and prolonged PT and higher lactate levels, suggesting a correlation between the severity of the asphyxia and the severity of thrombocytopenia.
The mechanisms responsible for the decreased platelet count are not entirely clear. In a significant number of infants, severe perinatal asphyxia is associated with disseminated intravascular coagulation (DIC), thus providing a reasonable explanation for the thrombocytopenia in these cases. However, some asphyxiated neonates develop thrombocytopenia in the absence of coagulopathy and the mechanisms underlying the thrombocytopenia in these cases are less clear. Findings in a rabbit model suggest that shortened platelet survival following exposure to transient hypoxia might lead to thrombocytopenia [42]. In contrast, a recent study by Christensen and colleagues described the thrombocytopenia of asphyxia as likely hyporegenerative and as a separate entity from, but also co-occurring with, disseminated intravascular coagulation (DIC) [40].
Recently, total body cooling became a widely accepted intervention aimed at improving the neurodevelopmental outcome of neonates with moderate to severe perinatal asphyxia [43]. Multiple studies have described changes in coagulation and platelet function induced by hypothermia. Specifically, in response to hypothermia, there is an inhibition of platelet activation, adhesion and aggregation as well as changes in the platelets’ surface antigen composition that can lead to their rapid removal from the circulation [44–46]. Boutaybi and coworkers compared thrombocytopenia due to asphyxia alone versus asphyxia treated with total body cooling and found there to be a three-times higher incidence of thrombocytopenia in infants treated with hypothermia [41, 47]. Importantly, this increase was in infants with only mild to moderate thrombocytopenia [41, 47]. Characterization of the time course of thrombocytopenia between the two cohorts showed that patients with asphyxia treated with hypothermia reached their nadir platelet count later than those with thrombocytopenia of asphyxia alone (day 5 versus day 3) [41, 47]. Christensen et al. also showed a significant lengthening of bleeding times and closure times measured with the Platelet Function Analyzer 100® (both measures of platelet function and primary hemostasis) during hypothermia, followed by rapid normalization after rewarming. This study suggests that the platelet functional impairment in asphyxiated neonates undergoing therapeutic hypothermia is transient [48].
The dysregulation of coagulation, platelet function, and the development of thrombocytopenia induced by both asphyxia and therapeutic hypothermia manifests clinically with a higher incidence of bleeding and coagulopathy [49–51]. A few studies have aimed at determining the modifiable risk factors for bleeding during therapeutic hypothermia. One study using thromboelastography (TEG) as a comprehensive assessment of coagulation reported an increased risk of coagulopathy with hypothermia, which was associated with specific measures of TEG but not with platelet counts [52]. Two separate studies found that, while platelet counts below 130 × 109/L or below 100 × 109/L were associated with an increased risk of minor bleeding, coagulopathy and hypofibrinogenemia were associated with severe bleeding [49, 53]. Together, these studies suggest that thrombocytopenia in the setting of perinatal asphyxia and therapeutic hypothermia may put infants at increased risk of mild bleeding, but coagulopathy and hypofibrinogenemia may be more important in the development of severe hemorrhage.
Infections
The association of thrombocytopenia with neonatal bacterial sepsis has been described by several groups of investigators, although the reported incidence varies depending on the definition of thrombocytopenia. For example, Guida and collaborators defined thrombocytopenia as a platelet count <100 × 109/L, and observed it in 54% of sepsis episodes [54], while Manzoni et al. defined thrombocytopenia as a platelet count <80 × 109/L, and detected it in 17.2% of septic infants [55]. Whether infections with certain organisms are more likely to induce thrombocytopenia than infections with other types (i.e., Gram positive vs Gram negative organisms) is controversial [54, 55], but it appears that virtually any bacterial organism capable of causing sepsis in a neonate is also capable of inducing thrombocytopenia. In modern NICUs, late-onset sepsis is the most common clinical condition underlying severe thrombocytopenia in neonates [56]. If the infection is treated appropriately, the thrombocytopenia usually lasts an average of 6 days (range 1–10 days) [54, 57]. However, a number of neonates with sepsis develop severe thrombocytopenia that persists longer than 2 weeks [56], and sepsis accounts for approximately one-third of NICU patients who qualify as very-high platelet users (i.e., >20 transfusions) [58].
The etiology of thrombocytopenia in bacterial sepsis is an active area of research. Prior studies assessing platelet production in neonates with sepsis and/or NEC showed a modest (2–3 fold) elevation in Tpo levels, circulating MK progenitors and reticulated platelet percentages (a measure of newly released platelets) [59], suggesting primarily a platelet consumptive etiology rather than decreased production. Interestingly, however, neonates with Gram-negative sepsis had only modest increases in thrombopoiesis, despite having more severe thrombocytopenia and more severe illness. This suggested that the thrombopoietic response in neonates can be dampened during severe illness, reaching a state of “relative hypoproliferation,” defined as a less than two-fold increase in platelet production in response to consumptive thrombocytopenia [59].
Traditionally, sepsis-induced platelet consumption has been attributed to DIC, increased platelet aggregation, and adherence to the damaged endothelium with subsequent removal by the reticulo-endothelial system, and/or immune mediated platelet destruction. With new research evidencing a central role of platelets in innate and adaptive immune responses, it is likely that the sepsis-induced consumption of platelets is more complex than previously thought. Platelets express and/or release multiple immune mediators, including cytokines, chemokines, and receptors known to be required for the initiation of the immune response to infection [60, 61]. Andonegui and colleagues demonstrated the expression of TLR-4 (the LPS receptor) on platelets and showed that, in response to LPS binding, platelets accumulate in the lungs where they bind to adherent neutrophils [62]. Clark and colleagues studied this further and described that, once LPS stimulated platelets adhere to neutrophils in the lungs, they induce robust neutrophil activation and the release of neutrophil extracellular traps (NETS) used to ensnare bacteria [63]. Furthermore, a recent study revealed that bacteria like Escherichia coli and Staphylococcus aureus are able to induce apoptosis in platelets by producing alpha-hemolysin or alpha-toxin, respectively, both enzymes that induce rapid degradation of platelet Bcl-xL, an anti-apoptotic protein essential for platelet survival [64]. This in turn results in a rapid drop in platelet count.
Viral and parasitic infections also can cause thrombocytopenia. The specific mechanisms of thrombocytopenia in these infections are unclear, but likely involve a mixture of platelet destruction and suppression of platelet production. Cytomegalovirus (CMV), the most common cause of congenital viral infection, affects megakaryopoiesis by directly infecting bone marrow stromal cells [65] and MKs [66]. In addition, CMV-infected infants frequently exhibit splenomegaly and require repeated transfusions due to platelet sequestration in the spleen. In regard to other viruses, herpes viruses can cause suppression of MK colony formation in vitro [67]. Parvovirus B19 usually causes severe anemia leading to hydrops fetalis, but it can also cause thrombocytopenia. In vitro, parvovirus B19 suppresses MK colony formation [68], and Forestier and coworkers found thrombocytopenia in 11 of 13 fetuses with parvovirus B19 infection [69]. Neonatal enterovirus infection can also cause thrombocytopenia, especially in the presence of hepatitis and DIC [70]. HIV has been recently recognized as a cause of thrombocytopenia in neonates. Like CMV and parvovirus, HIV can directly infect megakaryocytes and cause impaired platelet release, a phenomenon known as “ineffective platelet production” [71]. Adenovirus, Epstein–Barr virus, and dengue virus have also been reported to cause neonatal thrombocytopenia, although these are rather uncommon infections in the neonatal period.
Necrotizing Enterocolitis
Necrotizing enterocolitis (NEC) is frequently associated with thrombocytopenia. In an early study published in 1976 assessing hematological abnormalities in infants with NEC, as many as 90% of infants with NEC had platelet counts below 150 × 109/L, and 55% had platelet counts below 50 × 109/L [72]. Of the latter, 55% had bleeding complications, one-third of these serious enough to be considered contributory to the infant’s death. Six of 14 infants studied had evidence of DIC and the mean duration of thrombocytopenia was 7 days (range 1–31 days). A more recent study found that neonates with NEC and thrombocytopenia (platelet count <60 × 109/L) had two-times more bleeding events than neonates with similar degree of thrombocytopenia not related to NEC, but the bleeds were minor in severity [73]. In reviewing 58 cases of NEC treated in their institution, Ververidis and coworkers found that thrombocytopenia and/or a rapid fall in the platelet count was a poor prognostic factor, and that the degree of the thrombocytopenia correlated with the severity of the disease [74].
The etiology of thrombocytopenia in NEC is an area of active research. The shortened survival of platelet transfusions in these infants suggests a platelet-consumptive process. This was corroborated in studies of both murine models and human neonates with NEC/sepsis, which found markers of increased megakaryopoiesis in association with thrombocytopenia [59, 75]. However, a study by Cremer and coworkers found a reduced immature platelet fraction (IPF, a measure of platelet production; see Chapter 12) in severely thrombocytopenic neonates with NEC, suggesting decreased platelet production in these infants [76]. Interestingly, this subset of infants with NEC, severe thrombocytopenia, and low IPF had the highest mortality, suggesting that decreased IPF in the setting of severe thrombocytopenia could be a marker of illness severity and poor prognosis in NEC.
Platelets may also play an active role in the pathogenesis of NEC. Many pro-inflammatory cytokines are elevated in NEC, particularly platelet-activating factor (PAF), interleukin 1 (IL-1), and tumor necrosis factor (TNF) [77]. PAF, in particular, has been strongly implicated in the pathogenesis of NEC, based on animal studies demonstrating that the inhibition of PAF attenuates or prevents the development of NEC [78]. Interestingly, the levels of several pro-inflammatory cytokines (IL-1, IL-6, IL-8, TNF-alpha) increase in the supernatant of platelet units during storage in the blood bank, prior to platelets being transfused [79–83]. While most of these pro-inflammatory factors are released from leukocytes present in the platelet suspension, and thus levels are markedly reduced when the products are leuko-reduced prior to storage, bioactive factors and cytokines are also released from the platelet granules during storage, which could potentially contribute to the pathophysiology of NEC [84]. In that regard, at least two studies have suggested an association between higher number/volume of transfusions in patients with NEC and increased morbidity (short bowel syndrome and/or cholestasis) [85] or mortality (30.3 vs. 6.0 transfusions per 100 infant days in infants with NEC who died vs. survived in an unadjusted analysis) [84]. Although the difference in number of platelet transfusions between infants who died and those who survived was no longer significant in the latter study after adjusting for birth weight and severity of illness, the results strongly suggested the potential for such association. Thus, platelet transfusions should be administered judiciously to infants with NEC (See Chapter 12 for more discussion on platelet transfusions).
Thromboses
Neonatal thromboses can occur either spontaneously (such as renal vein thrombosis or sagittal sinus thrombosis), or in association with an indwelling central catheter [86]. Thrombocytopenia is not a mandatory component of the clinical presentation of neonatal thromboses, but the presence of persistent thrombocytopenia with no clear etiology should raise suspicion for the potential of thrombus formation, particularly if predisposing factors are present (including perinatal asphyxia, congenital heart disease, maternal diabetes mellitus, polycythemia, dehydration, twin pregnancy, or presence of an indwelling catheter). In recent reviews, thrombocytopenia/anemia or both were present in 51% of cases of renal vein thrombosis [87], but the classic triad of gross hematuria, palpable kidneys, and thrombocytopenia was found in only 13% [88]. Thus, a high index of suspicion is required to make an early diagnosis. While arterial thrombosis is less frequent than venous thrombosis in neonates [89, 90], a recent study in adults with consumptive platelet disorders, specifically heparin-induced thrombocytopenia and thrombotic thrombocytopenic purpura, found that platelet transfusion increased the risk for arterial thromboses [91]. While this study did not include neonates, the findings suggest that further study is warranted in this population. Further discussion of pathogenesis, presentation and treatment of neonatal thromboses can be found in Chapter 19.
Disseminated Intravascular Coagulation/Malformations
Disseminated intravascular coagulation (DIC) is a thrombo-hemorrhagic disorder in which there is systemic activation of the coagulation system, simultaneously causing intravascular thrombi formation (compromising blood supply to organs) and depletion of platelets and coagulation factors, resulting in bleeding. DIC is not a primary pathological process, it is always secondary to an underlying disorder such as sepsis, trauma, vascular abnormalities, or hypoxic ischemic encephalopathy [92]. Laboratory parameters associated with DIC include thrombocytopenia as well as deficits in coagulation factors that prompt frequent transfusions of blood products. The thrombocytopenia in DIC is secondary to thrombin-induced platelet aggregation [93]. Neonates have an increased incidence of DIC compared to pediatric or adult patients [94–96] which has been attributed to their dynamically evolving hemostatic system after birth [97–99].
Thrombocytopenia is one of the features of the Kasabach–Merritt syndrome (KMS), also known as “hemangioma thrombocytopenia syndrome.” It is now clear that the vascular tumors associated with the KMS are not true childhood hemangiomas, but rather hemangioendotheliomas [100]. Typically, the thrombocytopenia of KMS is preceded by enlargement and hardening of the vascular tumor. The primary mechanism is local consumption of platelets and clot formation, but platelets that have been damaged within the vascular abnormality are also removed in the spleen. KMS is refractory to transfused platelets and, if administered, they can result in painful tumor engorgement [101]. As a result, platelet transfusions are only indicated for active bleeding or immediately prior to surgery [102].
Drug-Induced Thrombocytopenia
A few drugs administered to pregnant women have been reported to cause thrombocytopenia in the mothers and their fetuses. However, only a small percentage of mothers who receive the particular drug have thrombocytopenia, thus making the firm establishment of this association difficult. Nevertheless, quinine, thiazide diuretics, hydralazine, and tolbutamide have been implicated as causes of fetal/neonatal thrombocytopenia when administered to pregnant women.
In addition, the development of moderate to severe late-onset thrombocytopenia in a well-appearing infant in whom infection, NEC, thromboses and DIC have been ruled out should trigger suspicion for drug-induced thrombocytopenia. The literature regarding drug-induced thrombocytopenia in neonates mostly consists of isolated case reports. However, there is a significant number of medications known to cause drug-induced thrombocytopenia that are also commonly used in neonates, including antibiotics (penicillins, linezolid, ciprofloxacin, cephalosporins, metronidazole, vancomycin, and rifampin), ibuprofen, acetaminophen, famotidine, cimetidine, hydrochlorotiazide, phenobarbital, and phenytoin [103]. If a neonate develops thrombocytopenia shortly after one of these medications has been started, and if other (more common) etiologies of thrombocytopenia have been ruled out, removal of the suspected medication should be considered.
One of the medications most frequently associated with thrombocytopenia among hospitalized patients is heparin, particularly unfractionated heparin. Heparin-induced thrombocytopenia (HIT) is an immune-mediated response to heparin administration. The incidence of HIT among neonates has been found to range from 0% to 1.5% [104, 105]. The incidence is slightly higher in pediatric ICU patients, particularly among those receiving therapeutic doses of unfractionated heparin for the treatment of thromboses or following cardiac surgery [106]. Unfractionated heparin in prophylactic doses [105] and low-molecular weight heparin are rare causes of HIT in pediatric patients [106].
HIT is a clinicopathological syndrome, which means that both clinical and laboratory characteristics have to be fulfilled in order to make the diagnosis. These include the presence of heparin, a fall in the platelet count by 50% or more, the correct time of onset (approximately 5–10 days after the initial exposure to heparin), and the presence of heparin dependent antibodies. There are two types of antibody assays used to diagnose HIT: antigen assays, which simply determine the presence of antibodies directed against the HIT-specific epitopes on platelet factor 4 (PF4), and functional assays, which determine the platelet-activating properties of the antibodies in a patient’s serum. The latter represent the gold standard in HIT antibody testing, because they detect the clinically significant HIT antibodies. However, they are technically demanding and are only carried out by large laboratories. Furthermore, it is unclear whether these assays need to be modified for use in neonates.
If HIT is strongly suspected or confirmed, heparin therapy should be discontinued, and alternative anticoagulants such as lepirudin and argatroban should be considered to avoid the thrombotic manifestations of HIT. However, data regarding the use of these anticoagulants in the neonatal population is sparse [107], and therefore their potential benefits need to be carefully balanced against the potential risks. For more information on HIT in neonates and children, the reader is referred to the excellent review by Risch et al. [106].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


