Abstract
This chapter focuses on the recognition and management of hemolysis in newborn infants (). Some of the common hemolytic anemias of childhood first appear in the newborn period, while others do not present until several months of age, and a few rare hemolytic disorders occur only in the neonatal period. These variations in the age that hemolytic anemia first presents reflect differences in neonatal erythropoiesis, hemoglobin synthesis, and the metabolism of newborn erythrocytes. When approaching an infant with a potential hemolytic disorder, the first issue to be addressed is whether there is evidence of increased red-cell destruction and accelerated production. If yes, then the next question to consider is whether the cause of neonatal hemolysis is due to extracellular (acquired) factors or an intrinsic (genetic) red-cell defect. Acquired disorders are those that are immune mediated, associated with infection, or accompany some other underlying pathology. Inherited red-cell disorders are due to defects in the cell membrane, abnormalities in red blood cell (RBC) metabolism, or a consequence of a hemoglobin defect.
This chapter focuses on the recognition and management of hemolysis in newborn infants (Table 10.1). Some of the common hemolytic anemias of childhood first appear in the newborn period, while others do not present until several months of age, and a few rare hemolytic disorders occur only in the neonatal period. These variations in the age that hemolytic anemia first presents reflect differences in neonatal erythropoiesis, hemoglobin synthesis, and the metabolism of newborn erythrocytes. When approaching an infant with a potential hemolytic disorder, the first issue to be addressed is whether there is evidence of increased red-cell destruction and accelerated production. If yes, then the next question to consider is whether the cause of neonatal hemolysis is due to extracellular (acquired) factors or an intrinsic (genetic) red-cell defect. Acquired disorders are those that are immune mediated, associated with infection, or accompany some other underlying pathology. Inherited red-cell disorders are due to defects in the cell membrane, abnormalities in red blood cell (RBC) metabolism, or a consequence of a hemoglobin defect.
Table 10.1 Causes of hemolytic anemia during the newborn period
|
|
Notes: CMV, cytomegalovirus; RBC, red blood cell; SLE, systemic lupus erythematosus
Evaluation of a neonate for hemolysis must be considered in the context of normal newborn physiology. The RBC lifespan in term neonates (80–100 days) and in premature infants (60–80 days) is shorter than in older children and adults (100–120 days) [1]. The reason for the reduced RBC survival observed in newborns is not known, but it has been thought to be due to the many biochemical differences between adult and neonatal RBCs [2–4]. Increased oxidant sensitivity of newborn red cells and relative instability of fetal hemoglobin have been considered as possible causes for this shortened lifespan [5]. Of interest, however, are recent studies using biotin labelled adult and neonatal RBC demonstrated that red cell survival in neonates was not that different: neonatal RBC survival (54.2+/−11.2 days) versus adult RBC survival (70.1+/−19.1 days) [6, 7]. Moreover, the survival of labeled adult RBC in neonates was much shorter than the 120-day lifespan seen when infused into healthy adults. These studies provide evidence that the circulatory environment of infants may be an important determinant of RBC survival rather than just factors intrinsic to the neonatal RBC.
Laboratory Features of Neonatal Hemolysis
A common clinical presentation of hemolytic anemia in older children is anemia, reticulocytosis, and hyperbilirubinemia. In the newborn infant, however, because of the normal hematologic adaptations to extrauterine life, the degree of hyperbilirubinemia and reticulocytosis must be interpreted in terms of the values appropriate for gestational and postgestational age. Also, some tests used to assess RBC production and destruction in older children have limited usefulness in the newborn.
Evidence for Anemia and Increased Red Blood Cell Production
The normal hemoglobin (Hb) concentration at birth is 14–20 g/dl (mean approximately 17 g/dl), decreasing to a level of 11 g/dl over 3 to 4 months [2, 8]. This hemoglobin decrement, referred to as the physiologic anemia of infancy, occurs as part of the normal adaptation to extrauterine life. It is a transition from a hypoxic milieu with an elevated RBC mass to a well-oxygenated environment with excess oxygen-carrying capacity. In the presence of mild hemolysis (commonly seen with ABO incompatibility), the magnitude of anemia may be minimal and obscured by the physiologic anemia of infancy. In infants with more significant hemolysis, the extent of anemia also is a reflection of the infant’s capacity for increasing RBC production. In many congenital hemolytic disorders, it is not uncommon to observe worse anemia in infancy compared with later in childhood, presumably a reflection of less available marrow capacity in neonates.
As fetal erythropoiesis diminishes, the mean corpuscular volume (MCV) decreases from birth (100–130 fl) to one year of age (70–85 fl). The elevated MCV at birth has implications on how we define neonatal microcytosis and thereby recognize certain disorders, such as alpha-thalassemia, in newborns [2, 9].
The normal reticulocyte count of children and older infants is 1%–2%. The reticulocyte count in term infants ranges between 3% and 7% at birth, but this decreases to 1% to 3% by 4 days and to less than 1% by 7 days of age [2]. In premature infants, reticulocyte values at birth are higher (6%–10%) and may remain elevated for a longer period of time. With significant hemolysis, neonates usually demonstrate increased reticulocytosis. However, as noted above, because the physiologic anemia of infancy is a response to excess oxygen-carrying capacity, mild hemolysis might not be sensed as an oxygen deficit and, hence, no increased erythropoiesis or reticulocytosis may be observed. Nucleated RBCs are seen in newborn infants, but they generally disappear by the third day of life in term infants and in 7 to 10 days in premature infants. The persistence of reticulocytosis or nucleated RBCs suggests the possibility of hemolysis. Hypoxia, in the absence of anemia, also can be associated with increased release of reticulocytes and nucleated RBCs. In the presence of hemolysis, the bone marrow manifests erythroid hyperplasia, but examination of the bone marrow rarely is utilized to diagnose hemolysis in neonates.
The peripheral blood smear in many hemolytic disorders reveals abnormal red-cell morphology. In newborn infants, however, the peripheral blood smear can be misleading unless “normal” RBC morphology for this age group is appreciated. Morphological characteristics of RBCs are consistent with the normal state of relative hyposplenism in newborn infants [9]. Howell–Jolly bodies, target cells, siderocytes, and other bizarre forms can be seen in normal neonates [10], and this is even more marked in premature infants. Just as with many other characteristics of the neonatal RBC, the red-cell morphologic changes found in the newborn period disappear by 3 to 4 months of age.
Evidence for Increased Red Blood Cell Destruction
Elevated serum unconjugated (indirect) bilirubin concentration with normal liver function is a useful marker of accelerated red-cell destruction in children and adults. In neonates, however, the normal increase in serum bilirubin concentration must be considered when evaluating for hemolysis. Bilirubin production in neonates is greater than in adults because of the increased circulating red-cell mass at birth, the decreased survival of neonatal red cells, and the increased enterohepatic circulation of bilirubin [11]. In addition, there is transiently reduced glucuronyl transferase activity, which impairs hepatic bilirubin conjugation [12]. As a consequence of these phenomena, physiological hyperbilirubinemia normally occurs in newborn infants, and bilirubin concentrations as high as 12 mg/dl (peak at 4 days) are seen in term neonates, and levels up to 15 mg/dl (peak at 7 days) are observed in premature infants. However, in association with hemolysis, serum bilirubin concentrations exceed these physiological levels and clinical jaundice may appear before 36 hours of age. Moreover, since mild hemolysis can occur without anemia or significant reticulocytosis, excessive hyperbilirubinemia often is the only manifestation of neonatal hemolytic disease. The decision to monitor bilirubin levels and to evaluate for underlying hemolysis is based on the Bhutani risk curves of total bilirubin concentration as a function of age [13] . Beyond the first few days of life, other metabolic problems and breast feeding can cause hyperbilirubinemia and further confound the recognition of increased red-cell destruction.
Carbon monoxide generation in humans is from the breakdown of heme, and is transported in the blood as carboxyhemoglobin. Measurement of blood carboxyhemoglobin levels can be used as a marker of accelerated red-cell destruction [14]. Also, exhaled carbon monoxide from the lungs can be measured directly to document increased RBC destruction [15, 16]. A noninvasive instrument to measure exhaled carbon monoxide is commercially available and appears to be a reliable marker of increased breakdown of hemoglobin to bilirubin [17, 18]. A neonate with hyperbilirubinemia and increased carbon monoxide production needs close monitoring because the cause of the elevated bilirubin is due to ongoing destruction of RBCs. This infant needs further evaluation to determine the cause of hemolysis.
Elevated serum lactic dehydrogenase (LDH) also is a marker of accelerated erythrocyte destruction; however, it is usually hyperbilirubinemia that initiates a work-up for neonatal hemolysis.
Other tests are available to assess for hemolysis, but most have limited utility in newborns. Serum haptoglobin is an alpha-2 glycoprotein that reacts with free hemoglobin to form a complex that is removed by the reticuloendothelial system. Since synthesis of this protein is not stimulated by hemolysis or clearance of the hemoglobin–haptoglobin complexes, reduced levels of serum haptoglobin are a reasonable marker of intravascular hemolysis. In neonates, however, haptoglobin levels are low and normal child or adult values may not be achieved until several months of age [19]. Thus, the serum haptoglobin concentration is not a reliable parameter of hemolysis in newborns.
Acquired Hemolytic Anemias in the Newborn
Alloimmune Hemolysis
Maternal sensitization to fetal RBC antigens inherited from the father and the ensuing placental transfer of maternal antibodies directed against fetal RBC antigens is the most common cause of neonatal hemolysis (see Chapter 9). The spectrum of clinical problems ranges from minimal anemia and hyperbilirubinemia to severe anemia with hydrops. At one time, before prevention of Rh sensitization was possible, hemolytic disease of the newborn was responsible for more than 10,000 deaths annually in the USA [20], but now the overall incidence of alloimmune hemolysis has decreased dramatically. Nevertheless, hemolysis due to Rh D incompatibility does still exist worldwide in areas where immunoprophylaxis is not readily available. At our institution in California, we have observed hemolysis in several neonates whose mothers had resided in Mexico and did not receive immunoprophylaxis with previous pregnancies. Today, the majority of alloimmune hemolysis cases in neonates are due to ABO incompatibility and, to a lesser extent, due to sensitization to Kell, Duffy, Kidd, and other Rh antigens. Since alloimmune hemolysis is the most common cause of neonatal hemolytic anemia, testing for antibody on neonatal RBCs is a necessary initial step in the evaluation of any newborn with hemolysis. This is accomplished with the direct antiglobulin test (DAT), also known as the direct Coombs’ test, which detects the presence of antibody on RBC. Only after alloimmune hemolysis is ruled out should one consider the work-up for other causes of neonatal hemolytic anemia. The specific tests used to diagnose other causes of hemolysis are discussed later in this chapter.
Immune Hemolytic Anemia Due to Maternal Disease
Maternal autoimmune hemolytic anemia or lupus erythematosus during pregnancy may be associated with passive transfer of immunoglobulin G (IgG) antibody to the fetus [21]. The diagnosis is suggested by the presence of neonatal hemolytic disease, a positive DAT, absence of Rh, or ABO incompatibility, and antiglobulin-positive hemolysis in the mother.
Hemolytic Anemia Due to Maternal Drug Intake
Maternal drug intake can lead to hemolysis in the fetus and neonate. One such example is seen with dapsone (diaminodiphenylsulfone), which is used for the prevention and treatment of Pneumocystis carinii infection, leprosy, and a variety of dermatologic disorders. A dose-related hemolytic anemia is recognized as a complication of this drug therapy in normal adults and children. Neonatal hemolysis has occurred when mothers were taking dapsone during pregnancy [22].
Infection
Congenital infections due to cytomegalovirus (CMV), toxoplasmosis, or syphilis, as well as bacterial sepsis, can be associated with hemolytic anemia, and frequently some degree of thrombocytopenia also exists. In association with congenital infections, often there is hepatosplenomegaly and other stigmata such as cataracts and microcephaly. In cases of bacterial sepsis, both the direct and indirect bilirubin may be elevated. The mechanism of hemolysis is not defined clearly, but it is thought to be related in part to RBC sequestration and macrophage activation associated with infection. In addition, certain bacteria may produce enzymes (phospholipases, neuraminidase) that affect membrane lipids or alter the normal RBC glycoprotein surface structure. Neuraminidase cleavage of sialic acid residues from membrane sialoglycoproteins. results in a decreased surface charge and shortened red cell survival [23].
Microangiopathic (Schistocytic) Anemia
The presence of many fragmented RBC or schistocytes in the peripheral blood suggests that hemolysis is due to altered erythrocyte–blood-vessel wall interactions. Abnormalities of the placental microcirculation or macrovascular anomalies such as an umbilical-vein varix are rare causes of congenital schistocytic anemia [24]. Also, schistocytic hemolytic anemia occurs as a component of disseminated intravascular coagulation (DIC) and this is secondary to the deposition of fibrin within the vascular walls. When erythrocytes interact with fibrin, fragments of RBCs are broken off, producing fragile schistocytes that are removed by macrophages in the reticuloendothelial tissues.
Vitamin E Deficiency
Premature infants are endowed at birth with significantly less vitamin E than term infants. Unless supplemental vitamin E is provided, this deficiency state persists for 2 to 3 months. Vitamin E is an antioxidant compound vital to the integrity of erythrocytes; in its absence, these cells are susceptible to lipid peroxidation and membrane injury. One clinical consequence of vitamin E deficiency is that hemolytic anemia can occur in small premature infants (weighing less than 1,500 g) at 6 to 10 weeks of age [25, 26]. This hemolytic anemia, which is characterized by reduced vitamin E levels and increased RBC peroxide hemolysis, disappears rapidly following vitamin E administration. At one time, it was thought that vitamin E deficiency might contribute to the anemia of prematurity in a more general sense. In fact, in one study performed several years ago, premature infants given daily vitamin E (15 IU per day) had higher hemoglobin levels and lower reticulocyte levels than a control group not given the vitamin [25]. However, these vitamin E supplemented babies still had lower hemoglobin concentrations than term newborns, indicating that anemia of prematurity is caused largely by other factors, such as erythropoietin deficiency. Although it has become standard practice to administer vitamin E to all premature infants, some still may develop hemolytic anemia. Those with anemia, reticulocytosis and an oxygen requirement may benefit from additional vitamin E [27].
Infantile Pyknocytosis
Pyknocytes are abnormal, densely staining erythrocytes with a distorted, irregularly contracted appearance with few, irregular, short projections. Pyknocytes account for up to 2% of the erythrocytes in a term infant’s peripheral blood smear and up to 6% of the erythrocytes in a preterm infant’s peripheral blood smear [28]. Infantile pyknocytosis is an infrequently observed hemolytic anemia of variable severity. It was initially described by Tuffy and colleagues [29] and has been noted by several investigators since that time [30–38]. Clinical manifestations include hemolysis, pallor, jaundice, and, occasionally, hepatosplenomegaly. Laboratory features include anemia, reticulocytosis, and an elevation in aspartate aminotransferase (AST). Increased numbers of pyknocytes, blister cells and polychromatophilia are apparent on the peripheral blood smear. Infantile pyknocytosis is largely a diagnosis of exclusion. Similar morphology may be observed with other hemolytic disorders, G-6-PD deficiency [30], pyruvate kinase deficiency, and vitamin E deficiency [25]. Hereditary elliptocytosis and microangiopathic hemolytic disorders also may produce similar-appearing RBCs. These causes of hemolytic anemia should be ruled out in the evaluation of any infant with proposed infantile pyknocytosis.
Hemolysis Due to Hereditary Red Blood Cell Membrane Disorders
The RBC membrane structure has three major components: (1) a bilayer of phospholipids intercalated with molecules of unesterified cholesterol and glycolipids; (2) integral membrane proteins (antigens, receptors) that penetrate or span the lipid bilayer, interact with the hydrophobic lipid core, and are bound tightly to the membrane; and (3) a separate protein network that forms the membrane cytoskeleton (spectrin, ankyrin, and several other proteins) interacting with both the integral membrane proteins and the lipid bilayer. Abnormalities in the quantity and/or quality of these proteins and their interaction with each other are the cause of the two most common inherited RBC membrane disorders hereditary spherocytosis and hereditary elliptocytosis (Table 10.2). Numerous DNA mutations have been identified, and several excellent reviews of the advances in these diseases are available [39, 40]. Both hereditary spherocytosis (HS) and hereditary elliptocytosis (HE) are associated with neonatal hemolysis.
| Disorder | Gene | Protein | Inheritance |
|---|---|---|---|
| Hereditary spherocytosis | ANK1 | Ankyrin-1 | AD |
| SPTB | Spectrin β chain | AD | |
| SPTA1 | Spectrin α chain | AR | |
| SLC4A1 | Band 3 anion transport protein | AD | |
| EPB42 | Erythrocyte membrane protein band 4.2 | AR | |
| Hereditary elliptocytosis | EPB41 | Protein band 4.1 | AD |
| SPTA1 | Spectrin α chain | AD | |
| SPTB | Spectrin β chain | AD | |
| Hereditary pyropoikilocytosis | SPTA1 | Spectrin a chain | AR |
| Ovalocytosis Southeast Asian type | SLC4A1 | Band 3 anion transport protein | AD |
| Overhydrated hereditary stomatocytosis | RHAG | Ammonium transporter Rh type A | AD |
| Dehydrated hereditary stomatocytosis | PIEZO1 | Piezo-type mechanosensitive ion channel | AD |
| Familial pseudohyperkalemia | ABCB6 | ATP-binding cassette sub-family B member 6 | AD |
| Cryohydrocytosis | SLC4A1 | Band 3 anion transport protein | AD |
Table 10.3 WHO classificaton of glucose-6-phosphate dehydrogenase variants
| Variant type | Residual enzyme activity | Clinical findings |
|---|---|---|
| Class I | Severe deficiency <10% normal | Chronic hemolytic anemia |
| Class II | Severe deficiency <10% normal | Episodic moderate–severe hemolysis with oxidative triggers |
| Class III | Moderate deficiency 10%–60% normal | Episodic mild–moderate hemolysis with oxidative triggers |
| Class IV | Normal activity >60% | None |
Hereditary Spherocytosis
Hereditary spherocytosis (HS) is the most common hereditary hemolytic anemia among people of northern European background. In the USA, the incidence of the disorder is approximately 1 in 5,000 [41, 42]. In most affected families, HS is transmitted as an autosomal dominant trait, and identification of the disorder in multiple generations of affected families is the rule. Nearly one-quarter of all newly diagnosed patients do not demonstrate a dominant inheritance pattern, and the parents of these patients are clinically and hematologically normal. New mutations have been implicated to explain some of these sporadic cases. Also, autosomal recessive mode of inheritance accounts for some cases of HS and is clinically manifested only in homozygous or compound heterozygous individuals, often associated with severe hemolytic anemia [43, 44].
The hallmark of the disorder is the presence of spherocytes, RBCs that have become spheroid because of a loss of membrane surface (Fig. 10.1a). This is a result of inherited mutations in genes for membrane cytoskeleton proteins (α-spectrin, β-spectrin, ankyrin, band 3, protein 4.2). These genetic mutations lead to protein abnormalities that weaken the stability of the interactions between the cytoskeleton and membrane lipid bilayer, resulting in a loss of vertical integrity of the cell membrane. As a consequence, there is vesiculation, with loss of bits of the bilayer and, thereby, progressive loss of membrane surface area. As RBCs become more spherical, there is decreased flexibility and increased vulnerability to entrapment in the spleen, and this is where metabolic depletion and macrophage interactions lead to hemolysis (Fig. 10.2). Removal of the spleen allows spherocytes to have a near-normal lifespan, although the cytoskeletal defects and abnormal RBC shape persist.
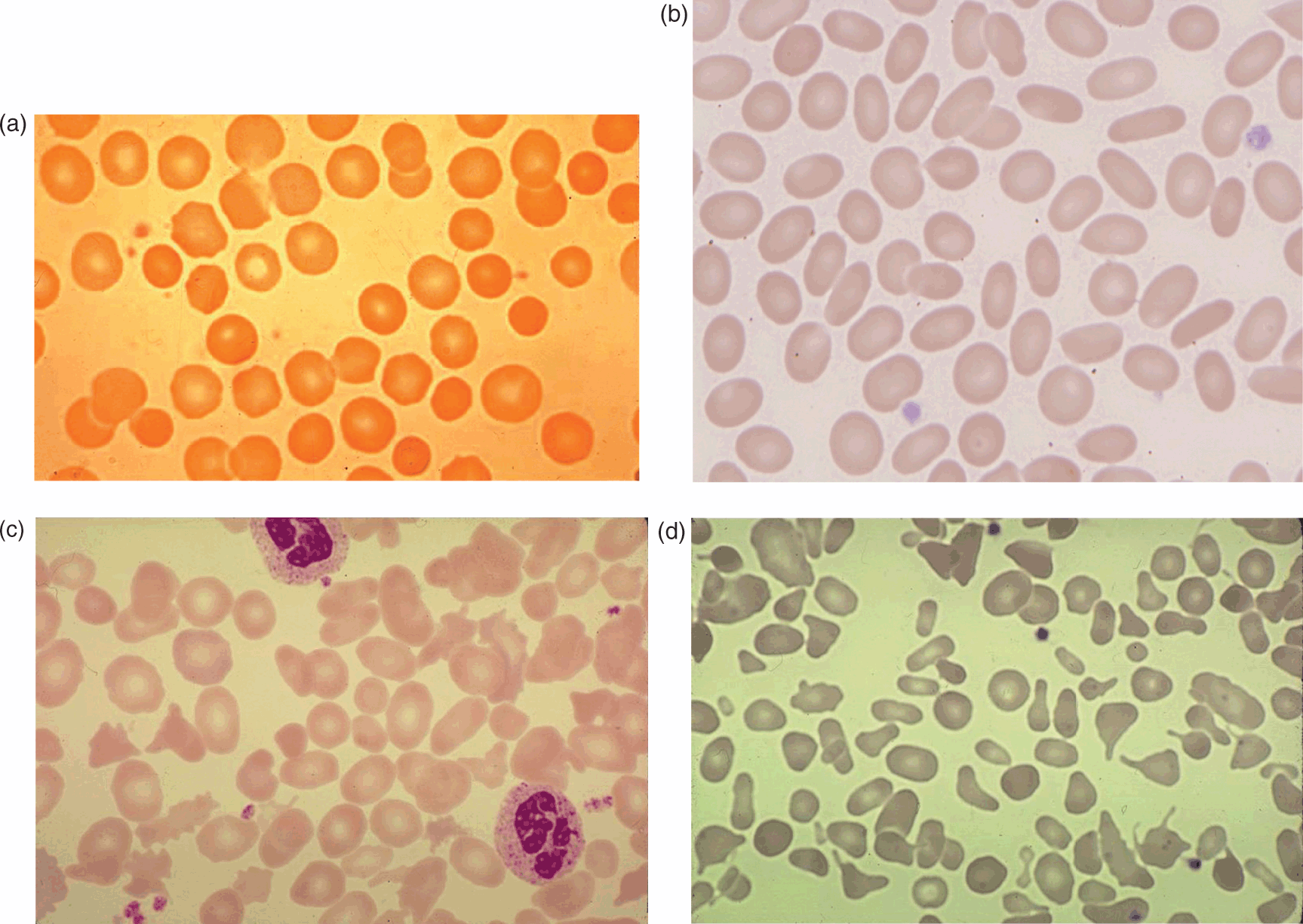
Fig. 10.1 Photomicrographs of blood smears from patients with different hereditary red blood cell (RBC) membrane disorders. (a) Hereditary spherocytosis; (b) mild, asymptomatic common hereditary elliptocytosis characterized by numerous elliptocytic cells; (c) hereditary elliptocytosis with chronic hemolysis with the presence of elliptocytes and rod-shaped cells and poikilocytes; (d) hereditary pyropoikilocytosis with a large number of microspherocytes and micropoikilocytes and relative paucity of elliptocytes.
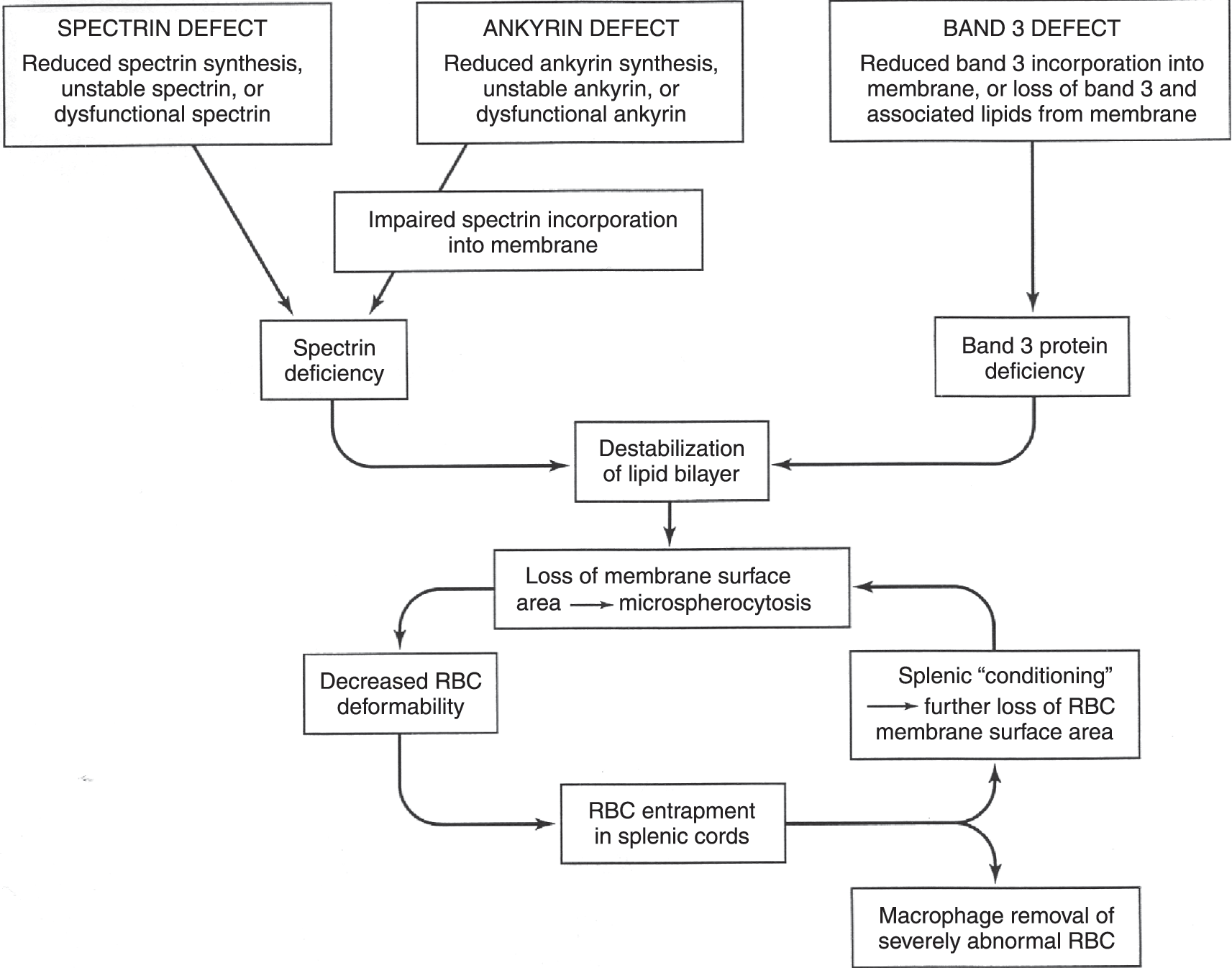
Fig. 10.2 The pathophysiology of hereditary spherocytosis is a consequence of spectrin, ankyrin, or band 3 abnormalities, which lead to membrane instability, loss of lipid microvesicles, decreased red cell surface area, reduced cellular deformability, and stagnation of red blood cells in the splenic cords.
The clinical features of HS encountered most commonly are anemia, jaundice, and splenomegaly. However, signs and symptoms are highly variable, with respect to both age of onset and severity. At one extreme, some mild cases may escape recognition for many years into adulthood, while, at the other extreme, hydrops with fetal death has been reported [40]. About 30%–50% of adults with the disorder have a history of jaundice during the first week of life [46, 47]. The magnitude of hyperbilirubinemia occasionally requires exchange transfusion [48]. In contrast to the frequency of hyperbilirubinemia, most newborns with hereditary spherocytosis are not anemic at birth [46]. However, in many infants hemolytic anemia does occur in the first month of life and can require one or more RBC transfusions. It is thought that this change reflects maturation of the splenic filtering function and development of the splenic circulation, coupled with an inadequate erythropoietic response. Within a few months, erythropoiesis increases, anemia improves, and the need for red-cell transfusions disappears in all but the most severely affected infants. Beyond the neonatal period, jaundice is rarely intense. However, even when patients have no detectable jaundice, there usually is laboratory evidence of ongoing hemolysis. Splenomegaly is common in older children, and, in large family studies, palpable spleens have been detected in over 75% of affected members. No apparent correlation exists between spleen size and disease severity.
The diagnosis of HS in older children is suggested by the complete blood count (CBC), which typically reveals decreased to normal haemoglobin concentration, increased mean corpuscular hemoglobin concentration (MCHC), increased reticulocytes, spherocytes on the blood smear (see Fig. 10.1a) and laboratory evidence of increased RBC destruction (increased bilirubin). In such an individual with a family history of documented HS, no further diagnostic investigation is warranted. In the newborn infant, morphologic assessment of the RBCs sometimes is difficult because even normal neonates may possess a minor population of spherocytes and, conversely, infants with hereditary spherocytosis may have fewer spherocytes than will be the case later in life. The possibility of HS in newborns is suggested by an increased MCHC (>36.5) and a low MCV. From the Intermountain Healthcare database, a neonatal HS ratio (MCHC/MCV), that is >0.36, indicates the presence of HS with a 97% sensitivity, >99% specificity, and >99% negative predictive value [49].
Historically, the incubated RBC osmotic fragility (OF) test was used to confirm the diagnosis of HS. However, this test lacks both sensitivity and specificity for HS and other conditions such as red cell glycolytic enzyme deficiencies can manifest increased fragility. In neonates, alloimmune hemolysis due to ABO incompatibility must always be ruled out since the peripheral blood spherocytosis and OF are similar to that seen in HS. Blood typing and a direct antiglobulin test (DAT) usually confirm a diagnosis of ABO incompatibility, although the DAT occasionally is negative and thus misleading. In the past, a practical approach in these cases was to repeat the OF testing in 3 to 4 months when the confounding effects of maternal antibody were no longer present.
The EMA (eosin-5-maleimide) binding assay is a flow cytometry test that provides a relatively rapid and automated screening tool for HS. EMA is a fluorescent dye which covalently binds to the extracellular portion of band 3 on erythrocytes. The underlying structural protein defects in HS RBCs result in decreased EMA binding which can readily be measured with flow cytometry [50]. This is a highly specific assay for HS and has become the diagnostic test of choice for many haematologists [51].
Molecular testing is indicated for patients without a classic family history and in those cases where the diagnosis is equivocal and might represent some other RBC membrane disorder [52]. This is important because splenectomy, which is appropriate for some older children with severe HS, may be contraindicated in other RBC membrane disorders which can look like HS [53].
Treatment during the newborn period is directed toward management of hyperbilirubinemia which usually requires phototherapy and/or exchange transfusion in severe cases. In affected infants who also are homozygous for the mutation responsible for Gilbert syndrome, hyperbilirubinemia almost always requires phototherapy [54]. Occasionally, RBC transfusions may be required at birth for management of symptomatic anemia [55]. A more common occurrence is the appearance of a transient but severe anemia during the first 20 days of life due to underproduction of erythropoietin in the face of continuing hemolysis [56]. These infants should be monitored closely following discharge from the nursery. Folic acid is required to sustain erythropoiesis: 0.5–1.0 mg per day is used for older children. With young infants we teach parents how to make an oral folate solution containing 50 μg folate/ml with 1 ml of a freshly prepared vitamin solution made each day. Splenectomy is the definitive treatment for severe hereditary spherocytosis. However, surgery is deferred until the child is at least 5 years old because of the increased risk of overwhelming sepsis with encapsulated organisms such as Haemophilus influenzae or Streptococcus pneumoniae that occurs in infants and young children following splenectomy [57]. It has been suggested that partial splenectomy may reduce the rate of hemolysis without increasing the risk of overwhelming infection, and this is a possible option in very young children with severe hemolysis [58].
Hereditary Elliptocytosis
Hereditary elliptocytosis is an autosomal dominant clinically heterogeneous group of disorders caused by mutations of RBC membrane cytoskeletal genes (usually spectrin or protein 4.1; see Table 10.2). These resultant protein abnormalities weaken skeletal protein interactions, disrupt the horizontal integrity of the membrane, and thereby increase RBC mechanical fragility [39, 40]. The hereditary elliptocytosis variants occur with an estimated frequency of 1 in 5,000, occurring in all racial and ethnic groups. Most individuals with common hereditary elliptocytosis have no clinical abnormalities or anemia, although the peripheral blood smear can be striking, with 15%–100% elliptocytes (see Figure 10.1b). In a small fraction (5%–20%) of common cases, there is some degree of hemolysis, usually compensated but occasionally leading to anemia (Hb 9–12 g/dl, reticulocytes 20%–25%). The peripheral blood smear in these hemolytic cases reveals poikilocytes in addition to elliptocytes. It is of interest that in families with common hereditary elliptocytosis, some individuals have a chronic hemolytic disorder while others do not, thus indicating that other genetic factors modify disease expression [59]. Both homozygous and compound heterozygous forms of the disease also occur and these cases are associated with clinically significant hemolytic anemia. Morphological characteristics include marked poikilocytosis, microelliptocytosis, and red-cell fragmentation (see Figure 10.1c). In some cases, both parents have nonhemolytic common hereditary elliptocytosis. Hereditary pyropoikilocytosis (HPP) is a recessively inherited severe hemolytic anemia characterized by RBC membrane budding, red-cell fragments, microspherocytes, and poikilocytes seen on the peripheral smear (see Figure 10.1d). The MCV may be extremely low (30–50 fl), while the MCHC is normal. From a clinical perspective, it is difficult to distinguish HPP from homozygous or doubly heterozygous common hereditary elliptocytosis. Once regarded as a separate condition, HPP is biochemically related to common hereditary elliptocytosis, occurs in families where other members have common hereditary elliptocytosis, and now is considered to be a variant of this disorder. During childhood, affected individuals manifest moderately severe hemolytic anemia (hemoglobin concentration 5–9 g/dl; reticulocyte count 13%–35%). Most but not all patients are of African heritage. The osmotic fragility test usually is normal in nonhemolytic forms of common hereditary elliptocytosis but is increased in those hemolytic variants with poikilocytosis and fragmentation. Molecular testing often is useful in these HE cases. No treatment is needed for most patients, although splenectomy later in childhood is beneficial for those with significant hemolysis.
Infantile poikilocytosis is a particularly interesting variant of hereditary elliptocytosis that occurs in the newborn period. Affected young infants have moderately severe hemolytic anemia and hyperbilirubinemia in the newborn period, the latter often necessitating exchange transfusion [60, 61]. The blood smear is characterized by marked red-cell fragmentation and poikilocytosis in addition to elliptocytosis. These morphologic changes are indistinguishable from those noted in patients with HPP. However, in contrast to HPP, hemolysis lessens gradually, and the clinical and hematologic features convert to those of mild hereditary elliptocytosis by 6–12 months of age [60]. In these infants, RBC membrane mechanical fragility is strikingly abnormal, probably a consequence of the destabilizing influence of large amounts of free intraerythrocytic 2,3-diphosphoglycerate (2,3-DPG) a byproduct of the presence of fetal hemoglobin [62]. As fetal hemoglobin levels decline in affected infants, membrane mechanical fragility improves, hemolysis disappears, and RBC morphology undergoes a transition from poikilocytosis to elliptocytosis. At birth, it is difficult to predict which patients will have transient poikilocytosis with ultimate recovery into common hereditary elliptocytosis and which are destined to have lifelong HPP with hemolysis. Close observation is necessary.
In addition to HS and HE variants, there are other rare RBC membrane defects than can present with neonatal hemolysis such as hereditary xerocytosis and the variable hereditary stomatocytic disorders (see Table 10.2). These are not discussed here but there are several excellent reviews of these conditions [39, 40, 53]. Nowadays, the diagnosis of these conditions is made by molecular testing panels after the more common RBC disorders are excluded [52].
Hemolysis Due to Hereditary Enzyme Abnormalities
Glucose is the main metabolic substrate for RBCs. It is metabolized by the glycolytic or “energy-producing” pathway and, to a much lesser extent, by the hexosemonophosphate (HMP) shunt or “protective” pathway (Fig. 10.3). The initial and most important reaction of the HMP shunt is catalyzed by glucose-6-phosphate dehydrogenase (G6PD). The HMP shunt represents the sole source of NADPH in erythrocytes, providing the reducing power necessary to protect RBCs from oxidant injury. The major products of glycolysis are adenosine triphosphate (ATP), a source of energy for numerous RBC membrane and metabolic reactions, and 2,3-DPG, an important intermediate that modulates hemoglobin–oxygen affinity. A critical enzyme for the generation of ATP is pyruvate kinase (PK). Hemolytic anemia due to G6PD deficiency is very common, while glycolytic enzymopathies are rare. Abnormalities in several glycolytic enzymes have been described, although over 90% of cases associated with hemolysis are due to pyruvate kinase deficiency. Hyperbilirubinemia, anemia, and even hydrops fetalis can be seen with inherited RBC enzymopathies. Overviews of this group of disorders are available elsewhere [63–65].
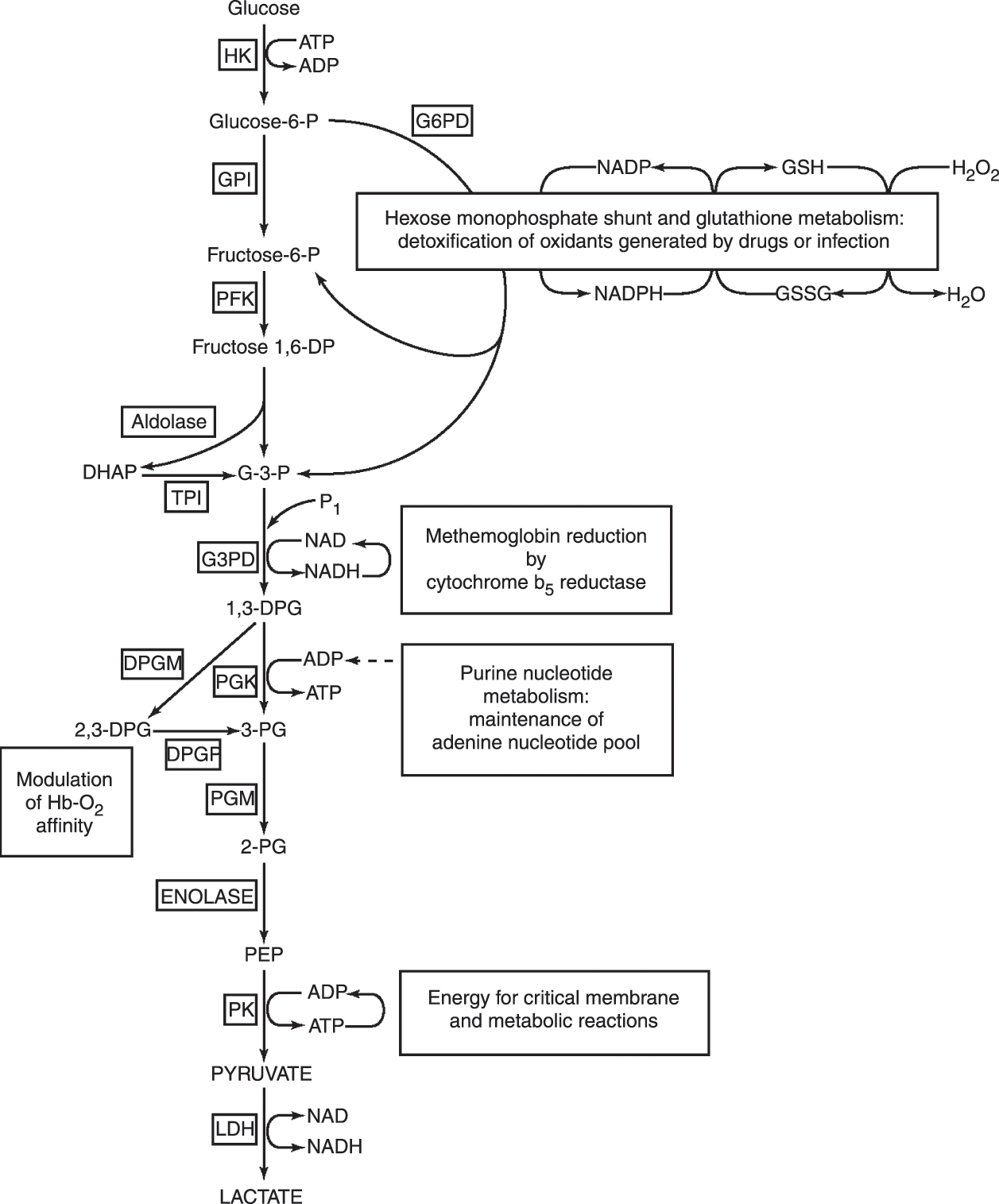
Fig.10.3 Overall red cell metabolism and function. Substrate abbreviations: (GSH) reduced glutathione; (GSSG) oxidized glutathione; (G-3-P) glyceraldehyde-3-phosphate; (DHAP) dihydroxyacetone phosphate; (1,3-DPG) 1,3-diphosphoglycerate; (2,3-DPG) 2,3-diphosphoglycerate; (3-PG) 3-phosphoglycerate; (2-PG) 2-phosphoglycerate; (PEP) phosphoenolpyruvate.
Cofactors: (ADP) adenosine diphosphate; (ATP), adenosine triphosphate; (NAD) nicotinamide adenine dinucleotide; (NADH) reduced nicotinamide adenine dinucleotide; (NADP) nicotinamide adenine dinucleotide phosphate; (NADPH) reduced nicotinamide adenine dinucleotide phosphate.
Enzymes are shown in boxes: (HK) hexokinase; (G6PD) glucose 6 phosphate dehydrogenase; (GPI) glucosephosphate isomerase; (PFK) phosphofructokinase; (TPI) triosephosphate isomerase; (G3PD) glyceraldehyde-3-phosphate dehydrogenase; (PGK) phosphoglycerate kinase; (DPGM) diphosphoglycerate mutase; (DPGP) diphosphoglycerate phosphatase; (PGM) phosphoglycerate mutase; (PK) pyruvate kinase; (LDH) lactate dehydrogenase.
Glucose-6-Phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked disorder that affects approximately 400 million people throughout the world, particularly in Mediterranean countries, Africa, and China. In the majority of G6PD-deficient individuals, there are no symptoms or laboratory signs of anemia in the steady state. Hemolysis occurs only with oxidant stresses during infections, after exposure to certain chemicals and medications, or associated with the ingestion of fava beans. Also, being an X-linked condition hemolysis occurs primarily in males and to a much lesser extent in carrier females.
Normal RBCs contain abundant amounts of reduced glutathione (GSH), a sulfhydryl-containing tripeptide that serves as an intracellular antioxidant, neutralizing peroxides that form during metabolism or are introduced directly from the extracellular environment. In contrast, G6PD-deficient RBCs have a limited capacity to regenerate GSH from oxidized glutathione, and in the absence of GSH, are vulnerable to oxidant injury (see Fig. 10.3). The effects of oxidants on RBCs are multifocal. Denatured globin precipitates, termed Heinz bodies, bind to the cell membrane, unfavourably altering its structure and function. Membrane lipid peroxidation may contribute to altered function. The ultimate result of these insults is hemolysis.
The clinical heterogeneity of G6PD deficiency is due to the very large number of different mutations, usually single amino-acid substitutions, that lead to altered enzyme function [67] (see www.bioinf.org.uk/g6pd/). The normal or wild-type enzyme is referred to as G6PD B, and many variant G6PD enzymes have been identified and characterized on the basis of biochemical properties. The World Health Organization (WHO) has further classified these different G6PD variants on the magnitude of enzyme deficiency and also the severity of hemolysis (Table 10.3) [68, 69]. Class I variant patients have severe enzyme deficiency, associated with chronic hemolysis, but these individuals are very rare. Class II variant patients also have severe enzyme deficiency (<10% activity). A common class II variant G6PDMediterranean, (563 C>T) found in those whose origins are in the Mediterranean area, the Near East and South Asia. For class II variants, oxidant-hemolysis may be quite severe and the hemoglobin may fall to life-threatening levels. Moreover, severe hemolysis can occur in children with class II G6PD variants following exposure to fava (broad) beans. Class III variant people have moderate enzyme deficiency (10%–60% of normal) with intermittent hemolysis, usually associated with infection or drugs. A very common variant in this class is G6PDA− (202 G>A/376A>G), found in 10%–15% of African Americans and with similar frequencies in western and central Africa. Hemolysis in children with class III variants usually is milder than class II and is self-limited.
Table 10.4 Drugs and chemicals associated with hemolysis in G6PD deficiency
| Unsafe (class I, II, and III G6PD variants) | |
|---|---|
| Acetanilid | Thiazolesulfone |
| Diaminodiphenyl sulfone (dapsone) | Phenazopyridine (pyridium) |
| Furazolidone (furoxone) | Phenylhydrazine |
| Glibenclamide | Primaquine |
| Henna (lawsone) | Sulfacetamide |
| Isobutyl nitrate | Sulfanilamide |
| Methylene blue | Sulfapyridine |
| Nalidixic acid (neggram) | Thiazolesulfone |
| Naphthalene (mothballs) | Trinitrotoluene (TNT) |
| Niridazole (ambilhar) | Urate oxidase (rasburicase) |
| Nitrofurantoin (furadantin) | |
In China, three major variants are recognized [70]. The most common is G6PD Canton (1376 G>T), which is usually reported to be a class II variant, although sometimes it is considered to be in class III. Another common variant is G6PD Kaiping (1388 G>A), which is usually classified as a class III variant although occasionally considered to be in class II. A third variant is G6PD Gaohe (95 A>G), which is usually considered to be a class II variant. These three variants account for over 70 percent of G6PD deficiency cases in China. The most common variant in Southeast Asia is G6PD Mahidol (487 G>A), a class III variant.
The recognition of G6PD deficiency in older children is suggested by the sudden onset of nonimmune hemolytic anemia associated with infection, fava bean ingestion, or the exposure to certain drugs and chemicals (see Table 10.4). Cells that appear as if a “bite” had been taken from them (due to splenic removal of Heinz bodies) and “blister cells” with hemoglobin puddled to one side may be seen on the peripheral blood smear. Although screening tests are available, definitive diagnosis requires assay of RBC G6PD activity. The diagnosis occasionally may be complicated in the setting of a high reticulocyte count, as is observed following a hemolytic crisis, because the population of deficient cells has been eliminated or, in transfused patients, because of the presence of normal, enzyme-replete RBC. Repeating the enzyme assay after at least 3 months ensures that any transfused cells are gone and that the population of deficient cells has been regenerated so that a more accurate determination of the presence of G6PD deficiency can be made. In some cases, specific G6PD mutations can be identified by DNA analysis, but this is used rarely in routine diagnosis.
Hemolysis resulting from G6PD deficiency is well documented in the newborn period [71], and may occur in utero [72]. However, the major consequence of G6PD deficiency in the newborn period is not hemolysis, but rather it is hyperbilirubinemia. Neonates with the rare class I variants are at greatest risk of neonatal jaundice, but these cases are rare. Most encountered infants with hyperbilirubinemia due to G6PD deficiency have one of the more common variants and come from the Mediterranean region or Asia, and to a lesser extent those with the G6PD A- variant. The degree of jaundice is quite variable; and in severe cases, there is a risk of bilirubin-induced neurologic dysfunction and kernicterus with permanent neurologic damage if the patient is not treated aggressively [73]. In neonates with Class II or III G6PD-deficiency, jaundice is rarely present at birth with the peak of onset occurring 2 to 3 days after birth. Jaundice is more prominent than anemia, which is rarely severe. Monitoring of jaundice and serum bilirubin levels in infants known to be G6PD-deficient is critical. [72, 74–76]. Of interest, African American infants with G6PD A− appear to be at lesser risk, although infants with the same G6PD variant in Africa have an increased incidence of neonatal hyperbilirubinemia [74, 77]. Moreover, in the latter group, untreated hyperbilirubinemia frequently leads to kernicterus with severe neurologic injury or death [77, 78]. Since black Africans and African Americans have the same G6PD A− genotypes, the adverse outcomes in Africa are thought to relate to local customs and differences in oxidant exposure [77, 78]. Herbs used in traditional Chinese medicine and clothing impregnated with naphthalene also are examples of covert oxidants to which susceptible infants may be exposed. Moreover, drugs (e.g., some sulfonamides), and fava-bean ingestion by mothers in late gestation have been implicated as the inciting stimulus in G6PD deficient newborns [79].
A study from Nigeria has reported a much poorer outcome for G6PD-deficient infants born at home, presumably a reflection of delayed identification and treatment of hyperbilirubinemia [77]. In the USA, there is concern that changes in healthcare delivery with early discharge of newborn infants may have similar consequences. In support of this is the report of four newborn infants with G6PD deficiency (three African American, one mixed Peruvian/Chinese) who developed kernicterus following early hospital discharge [80]. This report is disturbing because kernicterus in the USA has been rare in recent years and the adverse outcomes in these four cases occurred despite adherence to the early neonatal discharge guidelines of the American Academy of Pediatrics and the American College of Obstetricians and Gynecologists. Furthermore, in data from the USA Kernicterus Registry for the period from 1992 to 2004, over 30% of neonates requiring re-admission for hyperbilirubinemia and kernicterus were ultimately diagnosed with G6PD deficiency [73].
These disturbing findings have led some to suggest that universal neonatal testing programs for G6PD deficiency should be implemented [81]. Neonatal screening for G6PD deficiency has been very effective in reducing the incidence of favism later in life in Sardinia [82], and in other regions where this potentially fatal complication is common [71]. Thus, routine testing for G6PD deficiency is performed in many neonates with hyperbilirubinemia and/or those with less dramatic bilirubin elevations who are of Mediterranean, Nigerian, or East Asian ancestry. The American Academy of Pediatrics clinical guideline on the management of neonatal hyperbilirubinemia recognizes G6PD deficiency as a major etiologic risk factor for the development of severe hyperbilirubinemia and bilirubin-induced brain damage [83]. However, newborn screening for G6PD deficiency is only recommended for jaundiced newborns receiving phototherapy, whose family history, ethnicity, or geographic origin suggest the possibility of G6PD deficiency or for infants whose response to phototherapy is poor. Currently, in the USA the only newborn screening programs for G6PD deficiency are in the District of Columbia and the Pennsylvania newborn screening programs [84]. However, with changing immigrant populations, severe variants of G6PD deficiency nowadays are often found in many cities in the USA [85]. The need for point-of-care tests to be used on-site has been emphasized by various groups (e.g., prior to administration of antimalarial drugs). Similarly, for neonates, G6PD deficiency needs to be identified before an infant is discharged from the hospital. Since the maximum bilirubin does not occur until after 3 days of life, point-of-care enzyme testing in potentially affected infants is being assessed as a means of risk reduction for neonatal kernicterus [86].
In some cases, the hyperbilirubinemia seen in G6PD-deficient infants reflects accelerated red-cell breakdown, but more commonly there is no obvious RBC destruction or oxidant exposure. It has been suggested that hyperbilirubinemia may have another etiology, possibly related to impaired liver clearance of bilirubin. In support of this hypothesis are the observations that production of carboxyhemoglobin, a marker of hemolysis or RBC breakdown, is the same in G6PDMediterranean-deficient neonates with and without hyperbilirubinemia [87]. It is now thought that the variable degree of hyperbilirubinemia in G6PD-deficient neonates reflects the presence or absence of the variant form of uridine-diphosphoglucoronylsyl transferase responsible for Gilbert’s syndrome [67].
The major therapy for neonatal hyperbilirubinemia resulting from G6PD deficiency includes phototherapy or exchange transfusion to prevent kernicterus. RBC transfusion for symptomatic anemia, removal of potential oxidants contributing to hemolysis, and treatment of associated infections also needs to be addressed. In infants known to be G6PD-deficient, prevention of severe hyperbilirubinemia by administration of a single intramuscular dose of Sn-mesoporphyrin, an inhibitor of heme oxygenase, appears highly effective and safe, although this therapy has not been widely adopted [88].
Routine blood-bank screening likewise appears to be unwarranted, and G6PD deficiency is not considered a problem in transfusion medicine. Even in areas where G6PD deficiency is endemic, screening of blood donors is not required. Several years ago, a careful evaluation of the recipients of G6PD-deficient blood uncovered no deleterious consequences [89]. However, in premature infants, simple transfusions with G6PD-deficient red cells have been associated with hemolysis and severe hyperbilirubinemia requiring exchange transfusion [90]. Also, massive intravascular hemolysis has occurred in a South Asian neonate following an exchange transfusion with G6PD-deficient blood [91]. In view of these occurrences, it has been recommended that in areas where class II variants are common, donor blood should be screened for G6PD deficiency before transfusing premature infants or using the blood for a neonatal exchange transfusion. Currently, this recommendation is not standard blood-banking practice.
Pyruvate Kinase Deficiency
Pyruvate kinase deficiency (PKD) is an autosomal recessive disorder that occurs in all ethnic groups [64, 92]. Although it is the most common of the glycolytic pathway defects, it is rare in comparison to G6PD deficiency. Several hundreds of cases have been identified in Europe, Japan, and the USA [93]. The actual prevalence of PKD is somewhat in question with estimates ranging from 3 to 50 per million in the general white population [94–96]. Pyruvate kinase is one of the two key enzymatic steps that generate ATP in RBCs. Because nonerythroid tissues have alternative means of generating ATP, clinical abnormalities in pyruvate kinase deficiency are limited to RBCs.
The hemolytic anemia associated with PK deficiency is due to homozygous or compound heterozygous mutations in the PKLR gene located on chromosome 1 (1q21). More than 300 PKLR mutations have been identified [96, 97]. Reflecting this genetic diversity, the severity of hemolytic anemia in PKD varies considerably. Most PKD individuals are compound heterozygotes for two different PKLR mutations. A particularly high frequency exists among the Pennsylvanian Amish people, in whom the disorder can be traced to a single immigrant couple [98, 99].
Data from the PKD Natural History Study reveal that perinatal complications are common, occurring in 28% (65 of 233 births) and these include preterm birth, prenatal anemia requiring in utero RBC transfusions, intrauterine growth retardation, and even hydrops [93]. Approximately 90% of PKD patients (207 of 230 patients) have a history of neonatal hyperbilirubinemia which required phototherapy (93%) and/or exchange transfusion (46%). Jaundice tends to appear early (on the first day of life) and may require exchange transfusion. Beyond the neonatal period PKD is characterized by chronic hemolysis often requiring red blood cell transfusions. The severity of hemolysis and need for transfusion is minimized by splenectomy after 5 years of age.
The possibility of pyruvate kinase deficiency should be considered in any jaundiced newborn infant with nonimmune hemolysis in the absence of infections and without evidence of an RBC membrane defect or G6PD deficiency. RBC morphology in PKD is normal, although a few dense cells with irregular margins (echinocytes) occasionally are present. Assay of PK enzyme activity is the most useful and rapid way to make the diagnosis of PKD [100, 101]. Any case of PK deficiency detected by biochemical assay should be confirmed by molecular testing that is available in several referral labs. This is important because there is evolving evidence that the type of mutation relates to clinical prognosis [93].
Heterozygotes with one PKLR mutation have roughly half the normal amount of RBC PK enzyme activity, although sometimes it is hard to distinguish from patients with PKD. In all cases, however, PK heterozygotes are clinically and hematologically normal with no evidence of hemolysis.
Failure to demonstrate PK deficiency in the face of chronic hemolysis necessitates considering some of the other less common enzymopathies that occur in children (Table 10.5). Glucose phosphate isomerase (GPI) deficiency is the second most common glycolytic enzymopathy associated with hemolysis. The clinical manifestations of this disorder are identical to those of pyruvate kinase deficiency. The severity of hemolysis varies considerably. Anemia and hyperbilirubinemia complicate the postnatal course in many patients [102, 103]. Hydrops fetalis with death in neonates has been reported [104–106].
| Enzymopathy | Approximate fraction of enzymopathies | Mode of inheritance | Effects of enzymopathy |
|---|---|---|---|
| Hexokinase (HK) | <1% | AR | mild/severe CNSHA |
| Glucosephosphate Isomerase (GPI) | 3%–5% | AR | mod/severe CNSHA; +/− neurologic deficits |
| Phosphofructokinase (PFK) | <1% | AR | mild CNSHA; +/−myopathy |
| Aldolase | <1% | AR | mild/mod CNSHA; +/− myopathy |
| Triosephosphate isomerase (TPI) | <1% | AR | mod/severe CNSHA; neurologic deficits, |
| Phosphoglycerate kinase (PGK) | <1% | X-linked | mild/severe CNSHA; +/− neurologic deficits; +/− myopathy |
| Pyruvate kinase (PK) | >90% | AR | mod/severe CNSHA |
| Pyrimidine 5’nucleotidase (P5’N) | 2%–3% | AR | moderate CNSHA |
| Adenylate kinase (AK) | <1% | AR | CNSHA |
Phosphoglycerate kinase (PGK) deficiency is unique amongst glycolytic enzymopathies in that it is X-linked and most severely PGK deficient individuals with hemolysis also have neurologic abnormalities (seizures, mental retardation, aphasia, movement disorders) [107, 108].
Triose phosphate isomerase (TPI) deficiency associated with hemolytic anemia has been reported in several children. A unique feature of this enzymopathy is an early onset of a severe neurologic disorder characterized by spasticity, motor retardation, and hypotonia. Most affected patients die before they are 5 years old [109–111].
Hexokinase deficiency is a rare cause of hemolysis that has been identified in over 20 individuals [112, 113]. Splenectomy ameliorates but does not cure the hemolytic process.
Pyrimidine 5′nucleotidase (P5′N) deficiency is another rare enzyme abnormality associated with hereditary hemolytic anemia [114, 115]. Several kindred representing a wide geographic distribution have been reported, with a predisposition for people of Mediterranean, Jewish, and African ancestry. In all families studied, the disorder follows an autosomal recessive pattern. P5′N catalyzes the degradation of pyrimidine nucleotides to inorganic phosphate and the corresponding pyrimidine nucleoside [116, 117]. The most reasonable explanation for hemolysis in P5′N deficiency is that retained aggregates of ribosomes produce direct membrane injury, akin to that observed with Heinz bodies. There is mild to moderate anemia, reticulocytosis, and hyperbilirubinemia. RBC morphology is unique in that marked basophilic stippling is present. Basophilic stippling of RBCs is the morphologic equivalent of partially degraded ribosomes. Diagnosis requires a specific spectrophotometric enzyme assay.
Hemolysis Due to Hemoglobin Abnormalities
General Considerations
The earliest embryonic hemoglobins are composed of zeta globin chains (forerunners of alpha globin chains) and epsilon chains (forerunners of gamma globin and beta globin chains) (Fig. 10.4). The transition from zeta to alpha globin chains is complete by the end of the first trimester. Epsilon chains disappear more slowly and are replaced first by gamma globin and later by beta globin. Hemoglobin F (α2,γ2) is the hemoglobin found in fetuses after the first trimester, while Hb A (α2,β2) is the major hemoglobin in children and adults. At the time of birth, 60%–90% of hemoglobin found in normal term infants is Hb F. After birth, γ-globin synthesis declines as β-globin production increases, and by six months of age Hb F is approximately 5%. Only trace amounts of Hb A2 (α2,δ2) and gamma globin chain tetramers (γ4) are present in cord blood. With postnatal maturation, hemoglobin A2 increases gradually to the adult level of 2%–3.5%, while Hb Barts quickly disappears. In contrast to the major globin changes during development, heme is unchanged in structure in embryonic, fetal, and postnatal haemoglobins.
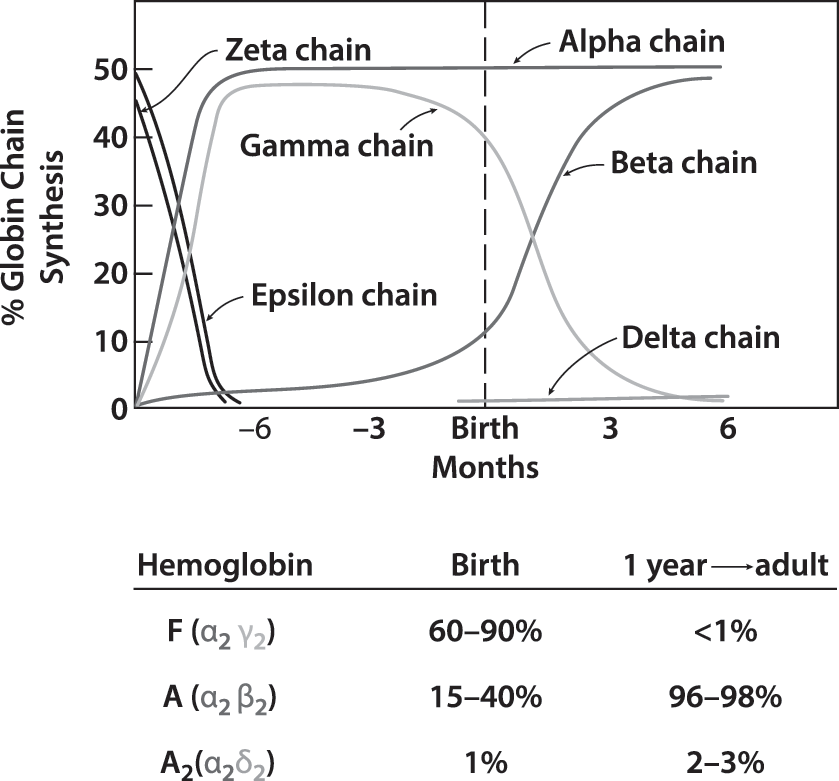
Hemolysis occurs with hereditary abnormalities due to decreased production of normal hemoglobins (thalassemia syndromes) or the production of qualitatively abnormal hemoglobins (sickle cell disorders, unstable hemoglobins). Those due to α-globin abnormalities are manifested at birth. In contrast, those due to β-globin abnormalities may not be apparent until four to six months of age when the switch from Hb F to Hb A synthesis reveals the defect. In contrast, rare gamma globin mutations are evident in fetal and neonatal life but disappear by three months of age when γ-globin synthesis is replaced by β-globin synthesis.
Thalassemia Syndromes
Thalassemia syndromes are characterized by diminished or absent production of normal α-globin polypeptides (α-thalassemia) or β-globin chains (β-thalassemia) [118]. Decreased globin synthesis causes a microcytic anemia, the severity of which depends upon the remaining number of functional α or β globin genes. However, since non-thalassemic globin chains are produced at a normal rate, the resulting imbalance of globin chain production also contributes to the pathophysiology. For example, in α-thalassemia, diminished synthesis of α-globin chains, leads to a relative excess of γ or β chains. In the fetus, excess gamma chains form tetramers (γ4), also known as Hb Barts, while beyond the neonatal period, the excess beta chains form tetramers (β4), also known as Hb H. The opposite is true of β-thalassemia in which α-globin chains accumulate, causing RBC membrane damage and hemolysis.
Alpha Thalassemia
Alpha thalassemia is of particular importance to neonatologists because its clinical manifestations are present in utero and at birth. These disorders occur worldwide, with an increased frequency in Africa, the Mediterranean, and throughout Southeast Asia, where the more severe forms of α-thalassemia are found. The genes directing α-globin synthesis (α2 and α1) are duplicated on the short arm of chromosome 16. Thus, with two genes per chromosome there are four genes, (α2α1/α2α1) that control normal alpha globin production. However, the α2 locus produces 2–3 times more globin than α1 does. Most α-thalassemia syndromes are due to deletions of alpha globin genes, although nondeletion α-thalassemia also occurs. Moreover, because nondeletion mutations involve α2, these thalassemia disorders are more severe [119–121]. Since the non-thalassemia globin chains are produced at a normal rate, gamma globin tetramers (γ4 or Hb Barts) and beta globin tetramers (β4 or Hb H) appear in substantial quantity as the number of functional alpha globin genes decreases. In families with α-thalassemia, an infant can inherit one, two, three, or four alpha thalassemia gene deletions, giving rise to the following four clinical alpha thalassemia syndromes: silent carrier state, α-thalassemia trait, hemoglobin H disease, or homozygous α-thalassemia (Table 10.6).
| Alpha thalassemia syndromes | Genotype | Clinical features |
|---|---|---|
| Normal | (αα/αα) | |
| Silent carrier | (−α/αα) | |
| Alpha thalassemia trait | (−−/αα) or (−α/−α) | Mild microcytic anemia |
| Hemoglobin H disease | (−−/−α) | Mild moderate hemolytic anemia |
| Homozygous alpha thalassemia | (−−/−−) | Severe anemia hydrops fetalis |
| Hemoglobin Constant Spring (Hb CS) syndromes | ||
| Heterozygous Hb CS | (αCSα/αα) | Similar to alpha thalassemia trait |
| Hb H disease with Hb CS | (αCSα/−−) | Similar to Hb H disease |
| Homozygous Hb CS | (αCSα/αCSα) | Similar to Hb H disease |
The silent carrier state
occurs in individuals lacking one functional alpha globin gene (−α/αα). In these cases, there are no clinical or haematological manifestations. At birth, the one distinguishing feature is a slightly increased concentration of Hb Barts (1%–3%) compared with that seen in normal neonates (less than 1%). Beyond the newborn period, there is no anemia or microcytosis, and the percentages of both Hb F and Hb A2 are normal.
Alpha thalassemia trait
is due to a deficiency of two α-globin genes. Deletion of two α-globin genes, in cis or trans, is associated with mild microcytic anemia, without hemolysis or reticulocytosis. Even neonates are microcytic (i.e., MCV less than 100 fl). The cord blood of infants with α-thalassemia trait usually contains 3%–10% Hb Barts. There are no significant clinical abnormalities associated with α-thalassemia trait, and beyond the newborn period this mild microcytic anemia can be mistaken for iron deficiency. In toddlers, the presumptive diagnosis of alpha thalassemia trait is made after iron deficiency, beta thalassemia trait, and hemoglobin E disorders have been ruled out. Definitive diagnosis is established by testing for the common α-globin gene mutations and/or α-globin gene sequencing. Of interest, alpha globin gene deletions occurring in Southeast Asians with α-thalassemia trait are in cis, or located on the same chromosome (−−/αα), whereas people of African descent with α-thalassemia trait have their gene deletions in trans, on different chromosomes (−α/−α). Consequently, the more serious alpha thalassemia disorders Hb H disease and homozygous α-thalassemia, (see below) almost exclusively are found in Southeast Asians and rarely in people of African ancestry.
Hemoglobin H (Hb H) disease
occurs in individuals with one functional α-globin gene. In Southeast Asia, it has been estimated that 13,000–16,000 infants with Hb H are born annually, translating into nearly 700,000 affected individuals living with Hb H disease. This disorder occurs most commonly when there is a deletion of three genes (−−/−α), in which case one parent usually has alpha thalassemia trait (−−/αα) while the other parent is a silent carrier for alpha thalassemia (αα/α−). Also, individuals who inherit the alpha thalassemia trait genotype on one chromosome and have a Hb Constant Spring gene on the other chromosome (−−/αCSα) also have hemoglobin H disease (see below). During the newborn period, Hb H disease is characterized by marked hypochromia, microcytosis, and increased concentration of Hb Barts (20%–30%). Beyond infancy, the imbalance in globin chain synthesis is associated with β-globin excess and the presence of Hb H. The latter appears as a rapidly migrating hemoglobin (5%–40% total Hb) on electrophoresis on cellulose acetate at alkaline pH or by other hemoglobin testing (HPLC, capillary electrophoresis). When erythrocytes from a patient with an intact spleen are incubated with the dye brilliant cresyl blue, small inclusions of precipitated Hb H are seen.
Infants with Hb H disease usually have minimal or no anemia; and beyond the newborn period there typically is a mild hemolytic anemia. Clinical features may include fatigue, and splenomegaly. Occasionally there are episodes of accelerated hemolysis in association with infections or exposure to oxidant agents. Parents of infants who have hemoglobin H disease should be instructed to avoid oxidant agents that can cause hemolysis (the same list that is given to patients with G6PD deficiency (see Table 10.4).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


