Central Nervous System Tumors in Children
Central nervous system (CNS) tumors account for 20% to 25% of all malignancies that occur in childhood. According to the North American Association of Central Cancer Registries (NAACCR), the age-standardized incidence rate was 48.47 per million in the 0- to 19-year age group for the period 2004–2007.1 The incidence was highest among children 1 to 4 years of age and lowest among 10- to 14-year-olds.
The etiology of pediatric CNS tumors remains largely unknown. Only 2% to 5% can be ascribed to a known genetic predisposition. Included in this category are those seen in patients with neurofibromatosis types 1 (NF-1) and 2 (NF-2), tuberous sclerosis, nevoid basal cell (Gorlin’s) syndrome, familial adenomatous polyposis, and Li-Fraumeni syndrome. An even smaller percentage can be attributed to ionizing radiation used for diagnostic or therapeutic purposes. For the majority of patients, no predisposing factors can be identified.
The management of children with CNS tumors has changed substantially over the past three decades. Routine use of magnetic resonance imaging (MRI), and now frequently also functional imaging, and improved neuropathologic examination and molecular diagnostics have contributed to better characterization of the different tumor types. Improved neurosurgical techniques and perioperative care permit greater degrees of surgical resection even for tumors previously considered inoperable because of their location in eloquent areas of the brain. All of these, as well as improved radiotherapy techniques, newer chemotherapy agents and regimens, and national and international clinical trials, have contributed to improved outcomes for children and adolescents with CNS tumors. According to the NAACCR, 5-year survival has increased from 62.9% for patients diagnosed in 1980–1989 to 75.3% for those diagnosed in 2000–2006.1
 RADIOTHERAPY FOR PEDIATRIC CNS TUMORS: GENERAL ISSUES
RADIOTHERAPY FOR PEDIATRIC CNS TUMORS: GENERAL ISSUES
Radiotherapy is an essential component of treatment for many children with CNS tumors. However, survivors are at significant risk for the development of long-term sequelae,2,3 many of which, while usually multifactorial, are in large part due to radiotherapy; many of the strategies used in the management of children with CNS tumors over the past three decades have been designed to reduce the risks associated with treatment. Recent developments in radiotherapy including improved targeting and new technologies and techniques for treatment, as well as new treatment modalities such as protons, all offer important opportunities for therapeutic gain that will be discussed below and in each section of this chapter.
Long-Term Effects of Radiotherapy
The quality of survival of children with brain tumors may be compromised by long-term sequelae. While some patients (e.g., patients with NF-1) may be at particular risk and while some sequelae (e.g., neurologic deficits) are more often due to the tumor and/or surgery, it is clear that radiotherapy is directly, alone or modulated by other factors, responsible for many late effects. A review by Kortmann et al.4 gives an excellent account of radiation-related sequelae in children treated for low-grade glioma including effects on brain parenchyma, neurologic deficits, neurocognitive and behavioral effects, endocrine dysfunction, vasculopathy, and the development of second tumors.
The neurocognitive sequelae of radiotherapy have become much better characterized over recent years. It is now known that myelinization and functional maturation of the CNS continue until well into adolescence and even into young adulthood. Through its effect on the microvasculature as well as on the oligodendrocyte precursor cells that produce myelin, radiotherapy causes disrupted neurogenesis and cortical atrophy. Patients fail to acquire new knowledge and skills at an age-appropriate rate and show a progressive decline in IQ over time.5 The magnitude of the deficit depends most importantly on age at treatment, but many other host (e.g., NF-1 or not), tumor (e.g., location, hydrocephalus or not), and treatment factors (e.g., radiotherapy volume and dose,6–8 use of chemotherapy9,10) play a role. Moreover, the development of other deficits such as behavioral difficulties related to the location of the tumor and/or surgery or hearing impairment due to cisplatinum may have a modulating effect. The end result for many patients is impaired school and social performance that deteriorates over time. There is increasing evidence that intervention using cognitive or behavioral therapy or pharmacotherapy and even exercise may be useful and that this should start soon after treatment for best results.5,11,12
Endocrine deficits are very common after radiotherapy.13 Even though a substantial proportion of patients may have had deficits prior to radiotherapy due to the tumor or to surgery,14,15 and even though there may be modulating factors such as chemotherapy that affect the frequency of deficits, radiotherapy is primarily responsible for the growth hormone deficiency that correlates with the dose of radiotherapy to the hypothalamic–pituitary axis16,17–18 and the primary hypothyroidism seen after craniospinal radiotherapy. There may be direct and indirect effects on musculoskeletal development. Osteopenia is a rather common finding that may put patients, particularly those with residual neurologic deficits, at significant risk for fracture.
Radiotherapy has been implicated as well in the development of cardiovascular complications including cerebrovascular events and coronary heart disease.15,19 Although again the etiology is likely multifactorial, it is important to be cognizant of the risks and to minimize the dose to vascular structures and the heart.
Strategies that have been used to avoid or minimize the long-term effects of treatment for pediatric brain tumors include the following:
• Avoidance of radiotherapy altogether (e.g., in patients with low-grade astrocytoma for whom surgery alone may be a good option)
• Delay of radiotherapy for young children (i.e., those younger than age 3 to 8) by the use of chemotherapy
• Use of daily anesthesia, improved immobilization techniques (e.g., rigid casts or a stereotactic frame), and/or daily pretreatment image verification, all of which allow the use of reduced safety margins
• Use of image-based treatment planning using computed tomography (CT)–MRI or CT–MRI–functional imaging coregistration and better treatment-planning and delivery techniques that result in greater sparing of normal brain and organs at risk
• Use of new radiation modalities (e.g., proton therapy that provides even greater sparing of the surrounding normal brain and organs at risk)
• Use of reduced radiotherapy target volumes when it is shown safe to do so (e.g., tumor bed rather than whole posterior fossa for the boost in standard-risk medulloblastoma)
• Reduction of radiotherapy dose (e.g., in young patients with standard-risk medulloblastoma for whom in the North American studies the dose for craniospinal irradiation has been reduced progressively from 35 to 36 Gy to 23.4 Gy and, in current studies, to 18 Gy for children younger than 8)
• Use of smaller fraction sizes where appropriate (e.g., 1.5 Gy/day for patients with radiosensitive tumors such as germinoma)
• Use of hyperfractionated radiotherapy (HFRT) (e.g., as in the current European studies for standard-risk medulloblastoma)
These will be discussed later in each relevant clinical situation.
Preparation for Radiotherapy
The planning and delivery of radiotherapy for children with CNS tumors are technically challenging and labor intensive for the entire interprofessional team. The expertise of specialist personnel such as pediatric nurses and play therapists can be pivotal in encouraging a young child to lie still for the making of an immobilization device, for radiotherapy-planning procedures, and for treatment itself. For children younger than age 4 or 5 years, daily anesthesia will almost always be necessary, and this will require a skilled pediatric anesthetist because anesthesia will be administered in an environment without all of the support available in an operating room.
Radiotherapy Target Volumes and Treatment Techniques
Focal, Tumor or Tumor Bed Radiotherapy
For most tumor types, target volume definition is best accomplished using CT simulation with CT–MRI image coregistration. For patients who have undergone cerebrospinal fluid (CSF) diversion or surgical resection or in whom tumor shrinkage has occurred with chemotherapy, it will be important to take into account any anatomic shifts that may have taken place. This will be more of an issue for tumors arising in some areas than others and often adds significantly to the time required for contouring. The clinical target volume (CTV) will be tumor type specific, while the planning target volume (PTV) will be technique specific, ranging from 1 to 5 mm depending on the type of immobilization device used and whether daily pretreatment image verification is to be performed.
Modern radiotherapy treatment-planning and delivery techniques make it easier to achieve conformity of the treated volume to the target and sparing of uninvolved normal structures than in the past. The choice of technique in an individual patient will require careful analysis of the dosimetry in the context of the available options. A recent article by Beltran et al.20 provides an excellent example of the issues to be considered now when weighing alternatives.
Other options for focal treatment include brachytherapy and intracystic injection of radioactive colloids. These will be discussed in the context of the relevant clinical situation.
Whole-Ventricle Radiotherapy
Whole-ventricular irradiation is used most frequently in patients with CNS germ cell tumors. Because subependymal spread is common, the target volume logically would include the lateral, third, and fourth ventricles with a margin of 1 to 1.5 cm. If lateral opposed fields are used, the volume of brain spared will be small. Better sparing can be achieved using intensity-modulated image-guided radiotherapy.21–23
Craniospinal Radiotherapy
The CTV for craniospinal radiotherapy has an irregular shape that consists of the whole brain and spinal cord and their overlying meninges. In standard techniques, the lower borders of lateral whole-brain fields are matched to the cephalad border of a posterior spine field, usually with a moving junction between the brain and spine fields to minimize the risk of underdose or overdose in the cervical spinal cord. Compensators may be needed to achieve dose homogeneity throughout the target volume.
Patient Positioning and Immobilization
Patients have traditionally received craniospinal irradiation (CSI) in the prone position, but modern technology allows safe treatment in the supine position that in general is more comfortable and, if anesthesia is required, allows better control of the airway. In either case, immobilization is essential and involves the use of a head shell or full-body immobilization. Careful attention to positioning at the time of simulation is critical to minimize or even eliminate the risk of certain long-term effects. For example, using neck extension together with careful selection of the level for the junction of the brain and spine fields, it is possible to avoid including the dentition in the exit from the superior aspect of the spinal field and thus damage to developing teeth.
Target Volume Definition
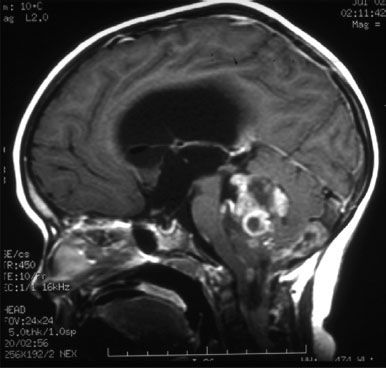
CT simulation is necessary to ensure adequate coverage of the CTV in the subfrontal region at the cribriform plate. Traditionally, blocks have been used in the lateral fields to shield not only the facial structures but also the lenses. However, in most children it is impossible to adequately irradiate the cribriform plate and shield the lenses (Fig. 84.1), and adequate PTV coverage should take precedence.
CT simulation is helpful, too, in identifying the lateral aspect of CTV for the spine field that includes the extensions of the meninges along the nerve roots to the lateral aspects of the spinal ganglia (Fig. 84.2). The field will be narrower in the dorsal region to avoid unnecessary irradiation of the heart and lungs and wider in the lumbar region, although here it is important to avoid an excessively wide field that will result in unnecessary irradiation of the bone marrow and gonads. The lower limit of the CTV for the spine field is best determined by MRI. Traditionally, the lower border of the spine field was placed at the lower border of the second sacral foramen, but it is well documented that the lower border of the thecal sac can be as high as L5 or as low as S3. In the interest of both CTV coverage and normal tissue sparing, it is important that the lower border be individualized according to the MRI findings.
FIGURE 84.1. The use of computed tomography simulation is superior to conventional radiographs for determination of the clinical target volume for craniospinal irradiation and ensures coverage of the meninges in the subfrontal region. In most children adequate coverage of the planning target volume precludes significant sparing of the lens.
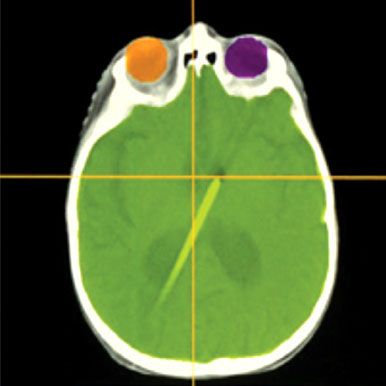
FIGURE 84.2. The use of computed tomography simulation with contouring of the cord and overlying meninges that extend laterally to the lateral aspect of the spinal ganglia results in a field width that is narrower than one based on bony anatomy. The addition of shielding further reduces the volume of normal tissues included in the treated volume.
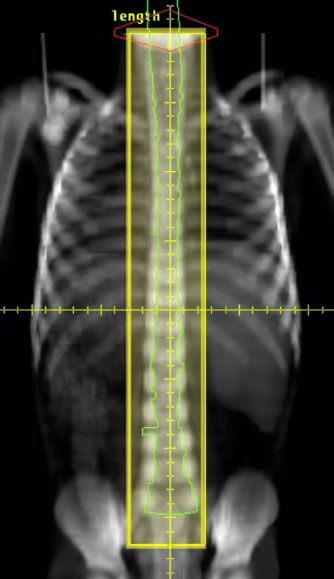
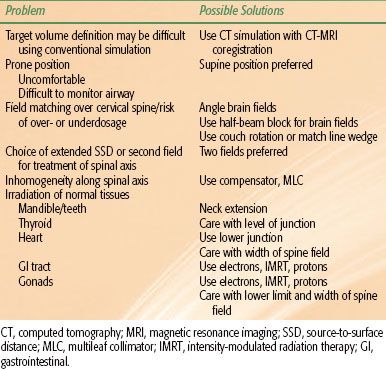
TABLE 84.1 TECHNICAL CONSIDERATIONS FOR CRANIOSPINAL IRRADIATION
Treatment Planning and Delivery
There are many issues that need to be addressed in designing a CSI technique (Table 84.1). Many of the different solutions24 add further complexity. Using modern tools for treatment planning and delivery, it is possible to greatly simplify the technique and substantially reduce planning and delivery times. One such technique is shown in Figure 84.3.25 In general, photons in the 6- to 10-MV range provide satisfactory coverage of the PTV. A variation of dose along the spinal axis of >10% will require the use of dose compensation that can be achieved using multileaf collimation.
Electrons are used in some centers to treat the spinal axis and in fact may be of greater interest now than in the past because of improved dose calculation algorithms and even electron dose modulation techniques. However, at the same time newer treatment-planning and delivery methods such as intensity-modulated radiation therapy (IMRT) together with daily image verification allow for improved dosimetry with photons with clinically relevant dose reductions to structures anterior to the target volume such as the heart, gastrointestinal (GI) tract, and gonads.26 The use of IMRT and smaller PTV margins raises new issues that are not as relevant when lateral opposed fields are used, such as the need to ensure adequate coverage of all CSF extensions including those along the optic nerves and into the internal auditory canals (Fig. 84.4).
New Treatment Modalities for CSI
Protons provide a dose distribution for CSI that cannot be achieved by even the most sophisticated photon beam treatment planning, with significant reduction in low-dose exposure outside the target volume.27 For now, however, access to proton therapy is limited and the cost prohibitive.
Radiation Dose and Dose-Fractionation Regimens
The conventional daily fraction size for the treatment of most pediatric CNS tumors is 1.8 Gy and the total dose typically on the order of 54 to 55.8 Gy. When treating a primary tumor of the spinal cord, it is conventional to use a lower total dose (e.g., 50.4 Gy). It is also usual to use lower doses for children younger than age 3 years to reduce the risk of neurocognitive deficits. When treating radiosensitive tumors such as intracranial germinoma, radiotherapy may be delivered using a lower dose per fraction (e.g., 1.5 Gy) and lower total doses of 30 to 45 Gy.
Many pediatric brain tumors exhibit a dose–response relationship for tumor control and in some cases local progression is not prevented by the use of a conventional “CNS tolerance” radiation dose. When the target contains only a small volume of normal brain tissue, dose escalation may be possible using newer treatment-planning and delivery techniques. HFRT may be a useful strategy in situations where dose escalation cannot be achieved safely using conventional fractionation.
FIGURE 84.3. To cover the clinical target volume for craniospinal irradiation, lateral opposed fields are used to treat the brain and a direct posterior field is used to cover the spinal axis. Magnetic resonance imaging is used to identify the caudal extent of the thecal sac. The field junction over the cervical cord should be at a level that avoids the inclusion of the teeth in the exit of the spinal field and usually is moved weekly (“feathered”) to avoid over- or underdosage. The supine position is more comfortable for the patient and safer if sedation or anesthesia is required. In the technique shown, fixed field parameters are used, which greatly facilitates treatment planning and delivery. (From Parker WA, Freeman CR. A simple technique for craniospinal radiotherapy in the supine position. Radiother Oncol 2006;78[2]:217–222.)
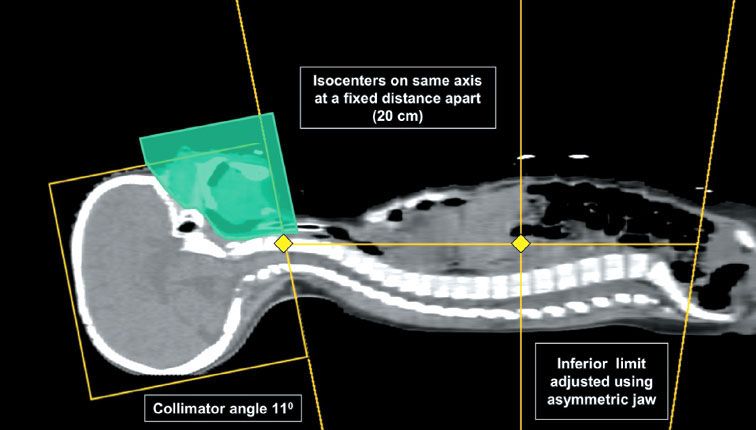
FIGURE 84.4. Postoperative axial T2-weighted magnetic resonance imaging for a patient receiving craniospinal irradiation (CSI) for medulloblastoma showing extension of cerebrospinal fluid along the optic nerves to the lamina cribrosa (A) and into the internal auditory canals (B). Care is necessary to identify all such extensions when using intensity-modulated radiation therapy for CSI given that the margins are much tighter than when using conventional lateral opposed fields.
TABLE 84.2 WORLD HEALTH ORGANIZATION CLASSIFICATION OF TUMORS OF THE CENTRAL NERVOUS SYSTEM

Follow-Up During and After Radiotherapy
An excellent review by Donahue28 describes the acute reactions seen during treatment with radiotherapy and provides guidelines for their management. These days nausea and vomiting almost always can be prevented by the use of the 5HT-3 antagonists. Headache is not an expected side effect and should be investigated by physical examination for signs of raised intracranial pressure and by imaging as appropriate. Steroids, if used, usually can be tapered by the second or third week of treatment. Fatigue is a rather common symptom and is cumulative. The neurologic status of the patient, especially coordination and gait, may appear to worsen during the last weeks of treatment because of this. Children usually recover relatively quickly and often can get back to their usual routine quite soon after completion of treatment.
Predictable effects of treatment include hormonal deficits, especially primary hypothyroidism when CSI is delivered using photons and growth hormone deficit secondary to inclusion of the hypothalamic–pituitary axis. Patients should be monitored closely in follow-up and treatment instituted as appropriate. Many will also need regular follow-up in ophthalmology and audiology.
Extra pedagogic support may be necessary. Patients should have ready access to a neuropsychologist for evaluation of any special needs and in the longer term to vocational assessment and counseling.
 RADIOTHERAPY FOR SPECIFIC TUMOR TYPES
RADIOTHERAPY FOR SPECIFIC TUMOR TYPES
The World Health Organization (WHO) classification of tumors of the nervous system29 is given in Table 84.2 and the distribution by tumor type and location in Figure 84.5. There are important differences between tumors seen in childhood and those occurring in adults. In children, almost half of all tumors arise in the infratentorial compartment. Low-grade astrocytic tumors as a group account for approximately one-third to half of all CNS tumors, but medulloblastoma is the most common distinct entity. High-grade gliomas, which account for the majority of primary brain tumors seen in adults, are much less common in children.
This chapter will follow the order of the WHO classification. Although in the past tumors arising in infants and very young children were grouped together and all managed similarly with chemotherapy in order to delay or if possible avoid radiotherapy, they are now managed according to the specific tumor type and so will be discussed here in each relevant section.
 ASTROCYTIC TUMORS
ASTROCYTIC TUMORS
According to the WHO classification, astrocytic tumors comprise the following clinicopathologic entities:
• Pilocytic astrocytoma (WHO grade I)
–Pilomyxoid astrocytoma
• Subependymal giant cell astrocytoma
• Pleomorphic xanthoastrocytoma
• Diffuse astrocytoma (WHO grade II)
–Fibrillary astrocytoma
–Gemistocytic astrocytoma
–Protoplasmic astrocytoma
• Anaplastic astrocytoma (WHO grade III)
• Glioblastoma multiforme (WHO grade IV)
–Giant cell glioblastoma
–Gliosarcoma
• Gliomatosis cerebri
These tumors are heterogeneous with respect to clinical presentation (age, gender, location in the CNS, imaging findings) as well as to growth potential and rate of progression. Two, pleomorphic xanthoastrocytoma and subependymal giant cell astrocytoma, are rare tumors. Their clinical presentation and imaging findings are quite characteristic and surgery is usually curative. They will not be discussed further here.
Low-Grade Astrocytoma (WHO Grades I and II)
So-called benign or low-grade astrocytomas (LGAs) comprise a heterogeneous group of tumors with behavior patterns that are fairly typical according to location and pathologic type. In general, in children, they follow an indolent clinical course with overall survival rates at 10 and 15 years as high as 80% to 100%. LGAs can be grouped according to their anatomic location:
• Cerebellar astrocytomas (15% to 20% of all CNS tumors)
• Hemispheric astrocytomas (10% to 15% of all CNS tumors)
• Midline supratentorial tumors, including the corpus callosum, lateral and third ventricles, and hypothalamus and thalamus (10% to 15% of all CNS tumors)
• Optic pathway tumors (approximately 5% of all CNS tumors)
• Brainstem LGAs (brainstem tumors account for 10% to 15% of all CNS tumors; 20% to 30% of these are LGAs)
• LGAs of the spinal cord (spinal cord tumors account for 3% to 6% of all CNS tumors; approximately 60% of these are LGAs)
Pilocytic astrocytomas are the most common type in the pediatric age group, accounting for almost all of the LGAs at certain sites (e.g., the cerebellum and the anterior optic pathway). They account for a smaller proportion of LGAs arising in the deep midline structures and in the cerebral hemispheres. Macroscopically, pilocytic astrocytomas appear well circumscribed and frequently have an associated cystic component. Pilocytic astrocytomas are characterized histologically by a biphasic pattern with a varying proportion of compacted bipolar cells with Rosenthal fibers and loose-textured multipolar cells with microcysts and granular bodies. Rare mitoses, occasional hyperchromatic nuclei, microvascular proliferation, and even infiltration of the meninges are compatible with a diagnosis of pilocytic astrocytoma and not a sign of malignancy. A variant, pilomyxoid astrocytoma, first described in infants and young children with chiasmatic/hypothalamic tumors, appears to be associated with more aggressive behavior that may include leptomeningeal seeding.30
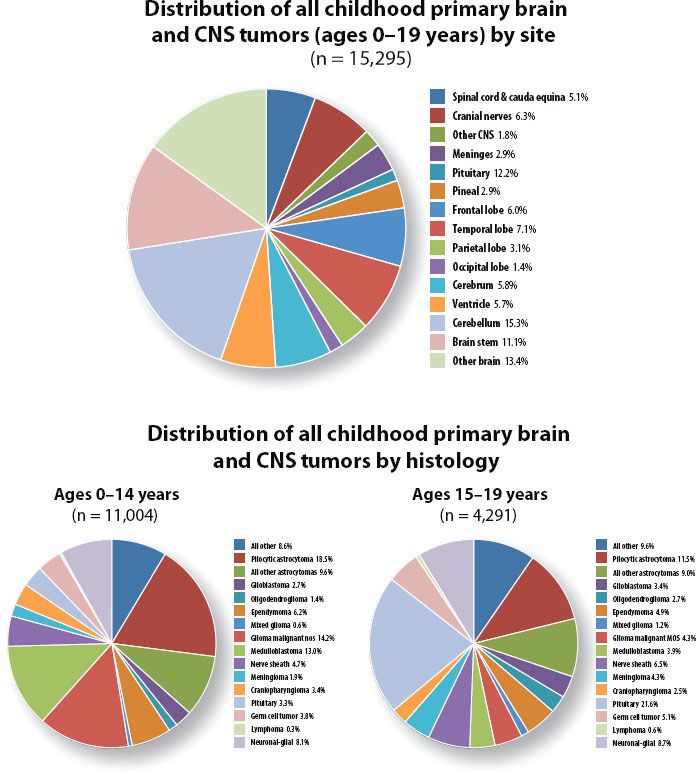
FIGURE 84.5. Distribution of central nervous system tumors by site (A) and histology (B) for the years 2004–2007. Data from the Central Brain Tumor Registry of the United States (www.cbtrus.org).
Diffuse astrocytomas account for only approximately 10% to 15% of all LGAs in children but for a relatively higher proportion of those seen in infants and adolescents. Most intrinsic pontine tumors and a large proportion of astrocytomas arising in the cerebral hemispheres are diffuse astrocytomas. Diffuse astrocytomas grow by infiltration rather than destruction of anatomic structures and usually are not well circumscribed. Microscopically, they are composed of well-differentiated fibrillary or gemistocytic neoplastic astrocytes on a background of loosely structured, often microcystic, tumor matrix. Cellularity is moderately increased. The presence of nuclear atypia is a diagnostic criterion, but mitotic activity, necrosis, and microvascular proliferation are absent. The growth fraction as determined by Ki-67 and MIB-1 labeling indices is usually low. Diffuse astrocytomas may undergo malignant progression, although this is not common in the pediatric age group with the notable exception of tumors arising in the pons.
Patients with LGAs typically present with a long history of nonspecific and nonlocalizing symptoms. Symptoms and signs of raised intracranial pressure may be seen in patients with midline and cerebellar tumors. Patients with posterior fossa tumors may present with neck stiffness and a head tilt as a manifestation of raised intracranial pressure causing tonsillar herniation, altitudinal diplopia, or spinal accessory nerve irritation. Seizures are present in as many as three-quarters of patients with hemispheric lesions. Other symptoms relatively less frequent and usually of more recent onset relate to the location of the tumor. These may include, for example, focal motor deficits with hemispheric tumors, visual field deficits with tumors compressing or involving the optic pathway, neuroendocrine deficits with hypothalamic tumors, and the diencephalic syndrome (consisting of emaciation with loss of subcutaneous fat despite normal or increased appetite, alert appearance, increased vigor and euphoria, pallor without anemia, and nystagmoid movements of the eyes) in young children with chiasmatic/hypothalamic tumors.
Neuroimaging findings are usually quite characteristic. Pilocytic astrocytomas are well circumscribed, often with a cystic component that may be large relative to the size of the solid component. There is usually little edema or mass effect. The solid component enhances brightly and uniformly with contrast material. Diffuse or fibrillary astrocytomas are usually not well seen on nonenhanced CT or MRI and usually show little enhancement with contrast material. T2-weighted or fluid-attenuated inversion recovery (FLAIR) MRI sequences usually best demonstrate the extent of disease.
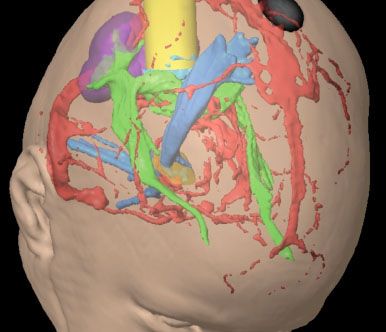
FIGURE 84.6. Surgical planning for a dysembryoplastic neuroepithelial tumor (light blue) in the right occipital lobe presenting with seizures. Functional magnetic resonance imaging data are used to identify the 1-degree visual cortex (purple). Fiber tracking identifies the optic radiations (green) and the corticospinal tract (darker blue). These data are then used to plan the trajectory (yellow). By identifying regions to be selectively avoided, similar information could be helpful in radiotherapy treatment planning.
Management of Low-Grade Astrocytomas: General Principles
Some children with LGAs may not require any tumor-specific treatment. These include, for example, patients with NF-1, as many as 15% of whom have optic pathway tumors.31 NF-1 patients also may have astrocytic tumors in other parts of the CNS as well as hamartomatous lesions, typically in the brainstem. These lesions may be detected on routine imaging, but even tumors that are symptomatic may remain stable over long periods so that surveillance is appropriate initial management.32–34 A number of clinical and imaging characteristics have been correlated with a more aggressive course, but even for these patients active intervention will usually be undertaken only at time of progressive disease that is symptomatic.
Progression-free survival without treatment may be very good as well for patients with tectal lesions who present with hydrocephalus without localizing brainstem signs. LGA in this region may be very indolent, showing either no progression (the majority) or only very slow progression over the course of many years following CSF diversion alone. The common characteristics of these very indolent tumors are their small size (<1.5 or 2 cm) and the fact that on imaging they are hypodense/hypointense and nonenhancing. Follow-up with MRI is essential to identify patients with progressive lesions as manifested by increasing size and/or enhancement with gadolinium. Treatment (usually radiotherapy but sometimes now surgery) at time of progression is associated with a high probability of long-term tumor control.
These special situations underscore the need for careful evaluation and individualization of management of patients with LGAs depending on the specific clinical situation and tumor type. If in doubt, a period of surveillance generally will be an acceptable initial approach.
Surgery is the mainstay of treatment for LGAs. Complete resection is more likely to be accomplished in patients with smaller tumors and those arising in noneloquent parts of the brain as well as in patients with the generally well-circumscribed pilocytic tumors. Modern surgical techniques that include, for example, computer-assisted resections (“neuronavigation”) aided by preoperative functional MRI, MR tractography, and intraoperative mapping of eloquent areas (Fig. 84.6) together with intraoperative MRI permit greater degrees of resection in larger proportions of patients, including many who in the past would have been considered to have inoperable lesions. Complete resection is now achieved in >80% of cerebral, cerebellar, and spinal cord tumors and about 50% of diencephalic tumors.
Children with LGAs who undergo complete resection fare very well, with long-term disease-free and overall survival rates of 80% to 100%.35–39,40 In most series, results are better (close to 100%) for patients with pilocytic astrocytomas than for those with diffuse or fibrillary LGAs, although some have disputed this, showing equally satisfactory results for both. In either type, postoperative adjuvant therapy is not indicated.
For children who undergo less than complete resection, the progression-free survival rate after surgery alone is less satisfactory. In a joint Children’s Cancer Group (CCG)–Pediatric Oncology Group (POG) study in which such patients were observed without adjuvant treatment, any residual tumor was associated with an inferior progression-free survival rate: the 8-year progression-free survival rate was 56% for patients with <1.5 cm3 residual tumor and 45% for those with >1.5 cm3. However, the majority of patients can be salvaged with a second surgical resection and/or radiotherapy. In the CCG–POG study, overall survival at 8 years was 95% and 90%, respectively.41
The role of postoperative radiotherapy following less than complete resection remains unclear. In most series, the use of radiotherapy in this situation results in improved disease-free survival without any benefit in terms of overall survival. Because only approximately half of all patients will develop progressive disease, the usual recommendation for a patient who is neurologically stable will be surveillance, with MRI performed at least every 6 months for the first 3 years, the period during which risk of progression is greatest.42 A second surgical procedure would usually be considered at time of progression, and other treatment, either radiotherapy or chemotherapy, reserved for patients with progressive, inoperable disease (Fig. 84.7).
In the past, patients with deep midline and other tumors considered surgically inaccessible were treated with radiotherapy, often even without histologic confirmation of diagnosis. However, using modern neurosurgical techniques, it is feasible to resect surgically about half of these lesions (Fig. 84.8), and the overall strategy for these patients should now be as for patients with LGAs at other locations, albeit with the understanding that outcome is not as satisfactory. The 8-year progression-free and overall survival rates in the CCG–POG study for patients with midline chiasmatic tumors were 25% and 84%, respectively.41
When adjuvant therapy is indicated, the options include chemotherapy, particularly for infants and young children and for patients of all ages with NF-1 who are at greatest risk of developing neurocognitive, vaso-occlusive, and neuroendocrine sequelae of treatment. Complete responses to chemotherapy are not common, but overall response rates that include stable disease range from 70% to 100% and the use of chemotherapy has been shown to permit delay of radiotherapy by 2 to as many as 4+ years.43–46 The age limit below which chemotherapy should be used is controversial. It is likely that delaying radiotherapy for 2 to 3 years will be of benefit for a child younger than age 5 to 8. However, the benefit from a similar delay for an older child is less clear, particularly when any benefit may be offset by neurologic compromise from further tumor progression as well as the need for a larger radiotherapy target volume. Moreover, recent advances in radiotherapy practice that have the potential to reduce the risks of radiotherapy have led to a reassessment of the role of radiotherapy in LGAs and better acceptance of its earlier use even in very young children.
FIGURE 84.7. An algorithm for the management of patients with low-grade astrocytoma.
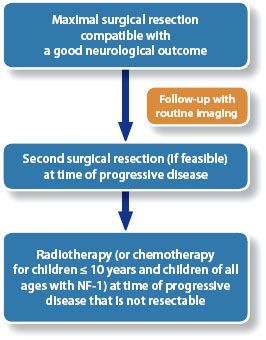
FIGURE 84.8. A 12-year-old boy referred to the radiation oncology department 3 years after diagnosis of a biopsy-proven JPA of the left thalamus at another institution. He had been deemed inoperable at diagnosis and had received chemotherapy first with vincristine and carboplatin and then with weekly vinblastine. A: At the time of referral he was having more frequent choreoathetotic movements of his right hand and there was evidence of progressive disease on magnetic resonance imaging (MRI). B: He underwent complete resection without complications and has no evidence of residual disease on his most recent MRI. This was a far better approach than radiotherapy, avoiding the neurocognitive, endocrine, and vascular complications associated with radiotherapy for tumors in this region, and with a greater probability of long-term tumor control.
Radiotherapy in LGAs
Due largely to improvements in surgery and to a lesser extent to successful treatment of younger children with chemotherapy, there has been a substantial decrease in the use of radiotherapy over recent years. Currently only approximately 10% of all children with LGAs receive radiotherapy.
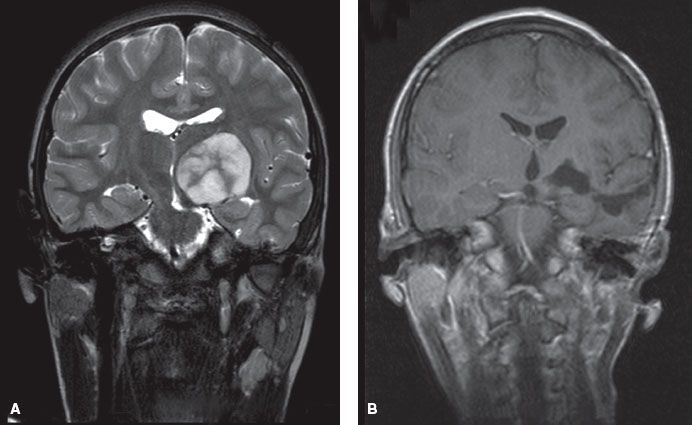
FIGURE 84.9. This patient underwent subtotal resection of a low-grade astrocytoma of the left cerebellar peduncle at age 8 (A: preoperative, B: postoperative). C: Three years later routine imaging showed evidence of progressive disease. The gross tumor volume for radiotherapy consists of the tumor as seen on magnetic resonance imaging at the time of treatment.
Indications for Radiotherapy
• Radiotherapy is not indicated after complete resection
• Radiotherapy may be indicated following incomplete resection in situations when tumor progression would compromise neurologic function (e.g., “threat to vision”)
• The clearest indication for radiotherapy is in patients with progressive and/or symptomatic disease that is unresectable
Radiotherapy Target Volume
The radiotherapy target volume (the gross tumor volume or GTV) consists of all disease seen on MRI performed just prior to treatment (Fig. 84.9). The resulting treatment volume usually will be considerably smaller than one based on preoperative imaging, which until recently would have constituted standard practice. Similarly, margins that were considered standard (and still are in adult practice) are unnecessary, particularly for the well-circumscribed pilocytic tumor for which a CTV margin of 1 cm40 or even 0 cm (i.e., CTV = GTV)47 around the GTV as seen on T1-weighted gadolinium-enhanced images has been shown to result in excellent local control. More generous margins of 1 to 1.5 cm around the GTV as seen on T2-weighted or FLAIR images may be more appropriate for the more infiltrative diffuse fibrillary tumors.
Radiotherapy Dose
Evidence for a dose–response correlation in LGAs in children is scant. Although the European and North American studies that randomized adult patients with LGAs between low-dose (45 and 50.4 Gy, respectively) and high-dose (59.4 and 64.8 Gy, respectively) radiotherapy failed to demonstrate any advantage for the higher dose, it may be unwise to extrapolate that in children doses of 45 to 50 Gy are as effective as the higher doses of 54 to 55 Gy that until now have constituted standard practice. There are biologic differences between LGAs in children and in adults. As well, children who have progressed on chemotherapy may have tumors that are less sensitive. For now, the recommendation for children with LGAs would be a “standard” dose of 50 to 54 Gy depending on the age of the child and the location of the tumor and its relationship to critical normal structures such as the optic chiasm.
Radiotherapy Technique
External-beam radiotherapy using a conventional dose-fractionation schedule should be considered the standard of care in LGAs and the technique to be used that which provides homogeneous irradiation of the CTV and best spares surrounding normal tissues.
Other approaches that have been used in LGAs include radiosurgery using the Gamma knife or linear accelerator–based techniques.48 While the use of a single fraction, typically of 10 to 20 Gy, to the periphery of the lesion or a small number of large fractions may not be optimal for diffuse or fibrillary LGAs with tumor cells embedded in rather than displacing normal brain, such treatment may be of interest for part or even all of the treatment for the less invasive pilocytic astrocytomas that are often small at time of progression.
Another option for treatment is brachytherapy, which has been used with some success particularly in European centers.49–53 As for radiosurgery, the principal limitation with respect to the use of brachytherapy is the size of the target volume. Brachytherapy series necessarily select for smaller tumors, and there is no evidence that brachytherapy is a better treatment option than external-beam radiotherapy. Radioactive solutions such as 32P, 90Y, 198Au, and 186Re may be useful in cystic LGAs, particularly for patients with recurrent disease after radiotherapy in whom symptoms not infrequently relate more to the cyst than to the solid component of tumor. Simple aspiration with or without placement of an internal shunt usually will alleviate symptoms for protracted periods, but in some cases, particularly those in which the cyst wall enhances and is felt to be biologically active, control of the cyst may be difficult, and the use of radioactive solutions could be considered. However, this is not a procedure without risks and care has to be taken to avoid leakage.
Follow-Up After Radiotherapy
Tumor regression after radiotherapy typically is slow and patients and their families need to be warned that the tumor may remain stable or sometimes even increase in size in the first months after completion of treatment. It is important not to assume that changes seen on MRI represent progressive disease. Close follow-up with early repeat imaging usually will be the best approach in this situation.
High-Grade Astrocytoma (WHO Grades III and IV)
Anaplastic astrocytoma (AA) is a diffusely infiltrating malignant astrocytoma characterized by nuclear atypia, increased cellularity, and significant proliferative activity. Glioblastoma multiforme (GBM) is the most malignant astrocytic tumor. Histopathologic features include nuclear atypia, cellular pleomorphism, mitotic activity, vascular thrombosis, microvascular proliferation, and necrosis. Both AA and GBM may develop from WHO grade II astrocytomas. However, with the exception of tumors that arise in the pons, they arise almost always in the pediatric age group de novo without evidence of a less malignant precursor lesion.
High-grade astrocytomas (HGAs) account for 5% of all CNS tumors in the pediatric age group. They are the most common tumor type in older adolescents. Two-thirds of HGAs are located in the cerebral hemispheres and the remainder approximately equally divided between the deep midline structures (thalamus and basal ganglia) and cerebellum. Patients with HGAs usually present with symptoms of short duration that relate to the location of the lesion.
Surgery is an important component of treatment. Because most studies show a survival advantage for patients who have undergone complete resection,54–58 maximal surgical resection compatible with a good neurologic outcome should be the goal for all patients and a second surgical procedure should be considered if there is significant residual tumor after the first. Postoperative radiotherapy always is indicated. Although leptomeningeal seeding is seen in a substantial minority (10% to 30%) of patients, the predominant failure pattern is local and the radiotherapy target volume is local, with a GTV that consists of the tumor bed and any residual enhancing or nonenhancing residual tumor plus any abnormality seen on T2-weighted MRI with a margin for the CTV of 1.5 cm. Because these are often large tumors and doses beyond tolerance levels for structures such as the optic chiasm are required, it is usual to consider a volume reduction at 50 to 54 Gy to a CTV that consists of the tumor bed and any residual tumor plus a reduced margin of 1 cm. The dose to the CTV should be at least 54 Gy given over 6 weeks, but a dose of 59 to 60 Gy is more usual if feasible. There is no evidence that higher doses delivered using radiosurgery or stereotactic boosts, boosts with brachytherapy, or HFRT result in improved outcome, but IMRT delivering accelerated treatment to a component of the target volume (such as the GTV) may be of interest, if only to decrease the overall treatment duration.
The role of chemotherapy remains to be defined. Although responses are seen to many different agents and regimens, results have often been difficult to interpret because of small patient numbers, inconsistent inclusion criteria with respect to pathology, and confounding variables such as tumor location and extent of surgical resection. The cooperative groups in North America and Europe are investigating a number of approaches that include newer chemotherapeutic and biologic agents, some of which have radiosensitizing properties. Whenever possible, patients should be treated on such protocols. Off study it may be difficult to make a recommendation with respect to adjuvant chemotherapy, although the poor prognosis, particularly for patients with macroscopic residual disease following surgery, usually is given as an argument for the use of the “current best” regimen, most often now temozolomide as in adults.
The prognosis for children with HGAs is poor, with a median time to progression of 10 to 11 months and an overall survival at 5 years of only approximately 20%. Several factors correlate with outcome. Patients with lesions in the cerebral hemispheres fare better than those with tumors in other locations, apparently independent of extent of surgical resection. The prognosis for children with thalamic lesions appears to be particularly poor.59 Age also may be an important factor. In contrast to most other tumor types, children younger than age 3 with HGAs fare better than older children, with overall survival at 3 to 5 years in the 33% to 50% range in the North American and United Kingdom Children’s Cancer Study Group/International Paediatric Oncology Society (UKCCSG/SIOP) baby studies.60–63 The prognosis may be even better for children younger than age 1.64 Histologic grade (AA vs. GBM) has not been shown consistently to affect outcome, but p53 overexpression and a high MIB-1 labeling index appear to identify patients with a particularly adverse prognosis.65
Management of Astrocytic Tumors in Specific Locations
Optic Pathway Gliomas
Optic pathway gliomas collectively account for approximately 5% of all CNS tumors in the pediatric age group. These are tumors of young children: the peak age incidence is between 2 and 6 years and 75% of all patients are younger than 10. One-third of patients have NF-1. They may be divided into three clinicopathologic entities: tumors confined to the optic nerve(s), tumors of the optic chiasm with or without optic nerve involvement (collectively “anterior” tumors), and tumors that involve the hypothalamus or adjacent structures (“chiasmatic/hypothalamic” or “posterior” tumors). Management of patients with optic pathway gliomas is often said to be controversial but is really not when differences in behavior between the different tumor types and between patients with and without NF-1 are taken into consideration.
Optic nerve gliomas may involve one or both optic nerves. Bilateral involvement is pathognomonic of NF-1. In a substantial proportion of cases, the optic nerve tumors are incidental findings on routine imaging and patients may remain asymptomatic with nonprogressive lesions over long periods; even spontaneous regression is well documented. The frequency of progression is difficult to establish, ranging from lows of <10% among patients followed in NF-1 clinics to 40% to 50% in series reported by oncology centers. Even in patients with symptomatic tumors, the course can be quite variable: only 30% to 60% of such patients will develop progressive disease that requires treatment.34,66 Thus, management of patients with optic nerve tumors will usually consist initially of close follow-up with regular ophthalmologic examinations and MRI, with active intervention, usually chemotherapy, reserved for patients with clear evidence of progression that is symptomatic.66–68
Patients with unilateral optic nerve involvement may not have NF-1. They present most frequently with proptosis that may be relatively long-standing. Findings on examination may include optic atrophy and impaired visual acuity. On MRI, optic nerve tumors are usually relatively small and well circumscribed, with bright enhancement typical for pilocytic astrocytoma. Biopsy is not necessary to make a diagnosis. Treatment usually, although not always, will be necessary and the approach will depend on whether there is useful vision. If not, then surgical resection will be the treatment of choice. If useful vision is preserved, chemotherapy would be the preferred option for infants and very young children up to age 5 and for patients of all ages with NF-1. Radiotherapy could be considered for older children. Overall, the prognosis is very good. Visual acuity remains stable or improves in the majority of cases following chemotherapy or radiotherapy. Long-term tumor control approaches 100% with either modality.69
Chiasmatic gliomas are tumors that involve the optic chiasm and sometimes one or both optic nerves as well. Patients typically present with loss of visual acuity and temporal field defects. On imaging, the tumors are usually relatively small and well circumscribed and enhance uniformly and brightly with contrast material, suggestive of pilocytic histology. Biopsy is usually not necessary.
A period of surveillance is appropriate initial management, particularly for patients with NF-1. For patients without NF-1, especially those who present before age 5, there is a high probability of early progression and the majority of patients will require treatment within a few months following diagnosis. Surgery is rarely an option for tumors in this location. As for optic nerve tumors, chemotherapy usually will be the treatment of choice for infants and young children and for patients with NF-1. Radiotherapy is reserved for salvage after chemotherapy and for definitive treatment of older children without NF-1, providing a reasonable expectation that vision will not deteriorate further and a probability of long-term progression-free survival in the 60% to 90% range.68,70–73 Overall survival for patients with chiasmatic tumors is in the 90% to 100% range.
Posterior, or chiasmatic/hypothalamic, gliomas account for approximately 70% of all optic pathway gliomas in children. They are typically rather large lesions that probably arise in the optic chiasm and extend to involve the hypothalamus. They may extend posteriorly along the optic tracts as well. They often fill the third ventricle, eventually causing hydrocephalus. Early findings consist of nystagmus, impaired visual acuity, and visual field deficits; only later do patients present increasing head circumference and/or symptoms and signs of raised intracranial pressure.
Treatment consists of CSF diversion, if necessary, and surgical resection, particularly if tumor is growing exophytically into the basal cistern because this may provide rapid relief of symptoms. In most patients, however, resection will be incomplete. As for LGAs at other locations, a period of surveillance following surgery is reasonable, although most patients will require adjuvant therapy.44,69 The treatment of choice for children younger than 5 and those with NF-1 usually will be chemotherapy. Progression-free survival at 3 to 5 years is rather low at 20% to at best 60%.44–46,74 However, some patients will never need further treatment, and for those who do, the use of chemotherapy allows radiotherapy to be deferred by a median of 2 to 4+ years without jeopardizing overall survival.
The indications for radiotherapy are (a) progressive disease on chemotherapy for children younger than 10 and (b) progressive disease at diagnosis or after surgery for older patients. Radiotherapy in this situation, given to local fields to a dose of 45 to 50 Gy for younger children and of 50 to 54 Gy for those older than 5, results in local tumor control in 70% to 80% of cases.44,69,73,75,76 Overall, however, outcomes are less satisfactory for this group of patients than for those with anterior tumors. Long-term survival is in the 50% to 80% range, and many patients will be left with significant neuroendocrine and neuropsychologic sequelae. Patients with NF-1 are particularly at risk. They may have subnormal IQ even without chemotherapy and/or radiotherapy.77 They are also at greater risk for moyamoya syndrome, a progressive vaso-occlusive process involving the circle of Willis. This may be seen without radiotherapy, but when radiotherapy is used in patients with NF-1 it is important to include MR angiography as part of the regular follow-up imaging protocol and intervene surgically if necessary to avoid a cerebrovascular accident.
Brainstem Gliomas
Tumors arising in the midbrain, pons, and medulla oblongata account for 10% to 15% of all CNS tumors in the pediatric age group. They are of several distinct types that can be broadly grouped as the more favorable low-grade focal, dorsal exophytic, and cervicomedullary tumors and the much more aggressive diffuse intrinsic pontine tumors.78,79,80
Focal tumors by definition are tumors of limited size (<2 cm) that on MRI are well circumscribed, without evidence of infiltration, and without edema. They may be cystic, and, as with cystic tumors at other sites, the cystic component may be large relative to the solid, biologically active, component. Focal tumors may occur at any level in the brainstem but most frequently are seen in the midbrain and medulla. They usually present with a long history of localizing findings such as an isolated cranial nerve deficit and a contralateral hemiparesis. Signs and symptoms of raised intracranial pressure are uncommon except in patients with tumors arising in the tectal region that may cause aqueduct stenosis while still small.
The management depends on the location of the tumor in the brainstem and the specific imaging characteristics of the tumor. As noted previously, patients with nonenhancing focal tumors in the tectal region who present with only hydrocephalus may do well without any treatment other than CSF diversionary procedures, usually endoscopic third ventriculostomy. Active intervention, including biopsy, is reserved for patients with clinical and radiologic evidence of progressive tumor. This is important because surgery for tumors in this location is associated with a substantial risk of morbidity even with modern techniques.
Surgery is the treatment of choice for focal tumors at other locations that are surgically accessible (meaning that they extend either toward the surface of the brainstem laterally or at the floor of the fourth ventricle) and have imaging characteristics suggestive of low-grade histology. In this regard, uniform bright enhancement with contrast material, which correlates with pilocytic histology, and the absence of peritumoral hypodensity are of particular importance. In experienced hands, the risk of morbidity for well-selected patients is low and, as with completely or subtotally resected LGAs at other sites, results may be excellent with freedom from progression in a majority of cases.81,82 Outcomes are less satisfactory for patients with bulky tumors and for patients with tumors in the medulla with lower cranial nerve deficits who are at risk of developing postoperative feeding and/or breathing difficulties.
There are several treatment options for patients with surgically inaccessible focal lesions. By extrapolation from series reporting results of treatment using conventional radiotherapy in brainstem tumors in which outcome correlates with location (pons vs. other), imaging appearance (tumor volume, density on CT, enhancement pattern), and histology (malignant vs. benign), it is reasonable to assume that 50% to 70% of focal lesions may be permanently controlled with such treatment. Similar results have been obtained in small numbers of patients treated with HFRT and with interstitial irradiation using 125I.49,51,53 There is also some experience with the use of radiosurgery. However, standard treatment is as for LGAs at other locations, that is, external-beam radiotherapy using a margin for the CTV of 0.5 cm, to a total dose on the order of 54 Gy given over 6 weeks. The risks of HFRT, radiosurgery, or stereotactic irradiation with large fraction sizes, or interstitial irradiation in inexperienced hands cannot be justified in the absence of any established superiority.
Dorsal exophytic tumors arise from the floor of the fourth ventricle. They are usually large, filling the fourth ventricle, but do not invade the brainstem to any significant extent. They present insidiously with failure to thrive in younger children and symptoms and signs of raised intracranial pressure in older patients. Cranial nerve deficits are seen in about half of the patients. On MRI, they are sharply delineated from surrounding structures. They are hypointense on T1-weighted images, are hyperintense on T2-weighted images, and enhance uniformly and brightly after gadolinium injection. Most are pilocytic astrocytomas.
Surgery is the treatment of choice for dorsal exophytic tumors. Intraoperative image guidance is essential to achieve a maximal degree of tumor resection. However, because there is usually no definite tumor–brainstem interface, even an optimal resection will leave a thin layer of tumor on the floor of the fourth ventricle. Nonetheless, the majority of children do well following surgery and routine postoperative adjuvant therapy is not indicated. Radiotherapy should be considered for the rare patient who is found to have a high-grade lesion or for patients with low-grade tumors who develop progressive disease in the early (<9 months) postoperative period. For patients whose tumors recur later, further surgery should be considered and radiotherapy reserved for those with inoperable disease. The radiotherapy volume and dose should be similar to those used for LGAs in other locations. The literature suggests that salvage is possible in the majority of cases and overall the prognosis for patients with dorsal exophytic tumors is excellent.83–85
Cervicomedullary tumors arise in the upper cervical cord and grow rostrally beyond the foramen magnum. Most are low-grade lesions whose axial growth is limited by the pyramidal decussations located ventrally at the junction of the cervical cord and medulla. At this point, the tumor grows posteriorly, causing a bulge in the dorsal aspect of the medulla, toward the fourth ventricle.78 These tumors typically present with lower cranial nerve deficits, sleep apnea and feeding difficulties in younger children, long tract signs, and sometimes torticollis. Hydrocephalus is unusual.
Surgery is the treatment of choice. Gross total resection may be achieved in 70% to 80% of cases and subtotal removal in most of the remainder. The probability of long-term tumor control after such treatment appears to be excellent for the typical low-grade lesion. There is, therefore, no indication for routine postoperative radiotherapy for these patients.
Diffuse intrinsic pontine tumors (DIPGs) account for 70% to 80% of all brainstem tumors. They arise in the pons and cause diffuse enlargement of the brainstem. Extension to the midbrain and medulla and/or exophytic growth is seen in at least two-thirds of cases. In contrast to the other types of brainstem tumors, the majority of DIPGs are fibrillary astrocytomas with a propensity for malignant change and a very poor prognosis.
DIPGs typically present with a short duration of symptoms consisting of multiple, bilateral, cranial nerve deficits (especially VI and VII) as well as long tract signs and ataxia. About 10% of patients have hydrocephalus at diagnosis. On CT, DIPGs are isodense or hypodense, with little enhancement after contrast injection, similar to diffuse fibrillary astrocytomas at other sites. They are best seen on T2-weighted or FLAIR MRI. The presence of ring enhancement is suggestive of high-grade histology.
Surgery has no role in the management of patients with DIPGs. Outside a clinical trial, even biopsy, a relatively nonmorbid procedure now, is considered unnecessary because in the context of a typical clinical presentation, the MRI findings are characteristic and histology does not influence treatment.86,87 Treated with conventional radiotherapy, the majority (70% or more) of patients with DIPGs will improve clinically. However, the progression-free interval is short (median <6 months) and survival is poor, with a median survival of <1 year and survival rates at 2 years <20%.
So far all attempts to improve the outcome for children with DIPGs have proved futile. HFRT was tested in a series of phase I/II studies using doses ranging from 64.8 to 78 Gy. Time to progression and overall survival were not improved in comparison with conventional radiotherapy.88,89 Moreover, at the higher doses of HFRT of 75.6 Gy and 78 Gy, morbidity was considerable. This included steroid dependency, vascular events, and white matter changes outside the radiation field, as well as hearing loss, hormone deficiencies, and late-developing seizure disorders in the small number of long-term survivors.88,90–92 Accelerated and hypofractionated radiotherapy regimens have also been tested in single-institution studies using, respectively, a total dose of 50.4 Gy given in 28 twice-daily fractions of 1.8 Gy over 3 weeks93 and 39 Gy in 13 daily fractions and 45 Gy in 15 daily fractions.94,95 Progression-free and overall survival rates were similar to those seen in the HFRT studies.
Alternative approaches that use chemotherapy in combination with radiotherapy also have been disappointing. None of the many single-agent and multiagent regimens that have been tested in this patient population have been shown to provide a survival advantage compared with radiotherapy alone. New agents and novel chemotherapy–radiotherapy combinations are under investigation by the pediatric cooperative groups in North America and Europe.
Currently, standard treatment for DIPGs consists of radiotherapy given to the GTV as usually best demonstrated on T2-weighted or FLAIR MRI with a margin for the CTV of 1 to 1.5 cm to a dose of 54 Gy given in 30 daily fractions over 6 weeks that because of the initial rapid progression of neurologic deficit treatment often needs to be started on a semi-urgent basis. Because no unexpected toxicity was seen with the hypofractionated regimens, these in some cases could be considered an alternative to conventional radiotherapy that reduces the burden of treatment for the child and family.
Improvement in clinical status is usually evident as early as 2 to 3 weeks into treatment and steroids, if used, usually can be discontinued at that point. This improvement is often impressive and well appreciated by the family, despite being of only short duration. Treatment at time of progression may include experimental chemotherapy regimens or supportive care, or even retreatment with radiotherapy in selected cases.96,97
Astrocytoma of the Spinal Cord
Intramedullary spinal cord tumors account for 3% to 6% of all CNS tumors in the pediatric age group. About 60% are astrocytomas, the majority of which are LGAs; 30% are ependymomas; and the remainder, gangliogliomas and developmental tumors such as teratomas, lipomas, and dermoid and epidermoid cysts.
Patients typically present with pain and motor deficits, often of long duration. Rapid progression of symptoms and signs is suggestive of high-grade histology. On imaging, astrocytomas most often are seen to comprise a solid component and one or more cysts that may be intratumoral and/or extend rostrally and caudally beyond the solid component. Most enhance heterogeneously with the use of contrast material. Ependymomas, in contrast, only rarely harbor intratumoral cysts (although they may have an associated syrinx) and usually enhance homogeneously.
The management of spinal cord tumors has changed over recent years.98 In the past, patients typically were treated with biopsy followed by radiotherapy, and there is evidence to suggest that this is effective treatment in more than half of children with LGAs.99 However, improvements in surgery and the routine use of surgical adjuncts such as ultrasonic aspiration and support systems such as intraoperative ultrasonography and sensory and more recently motor evoked potential monitoring improve the safety and completeness of resection so that complete or subtotal resection is now possible in approximately 80% of children with pilocytic astrocytoma (the majority), especially those with a syrinx. Resection is more difficult and less likely to be complete in patients with grade II astrocytoma because of the more infiltrative nature of these tumors and the absence of a clear interface.98 Because outcome following complete or subtotal resection for LGAs is very good, with long-term progression-free survival in the 70% to 90% range,100–102 routine postoperative adjuvant therapy is not indicated. For children in whom complete or subtotal resection is not possible, the options will be as for patients with LGAs in other locations, that is, early second surgery if feasible, or close follow-up with second surgery and/or radiotherapy at the time of progression. As for LGAs in other locations, chemotherapy may be an alternative to radiotherapy for young children.103,104 The benefit of surgical resection in HGAs is less clear and the more usual approach is biopsy followed by postoperative radiotherapy.105
The radiotherapy target volume for LGAs consists of the solid portion of the tumor (including intratumoral cysts) with a margin for the CTV of 1 to 1.5 cm. The usual dose is 50.4 Gy given in 28 daily fractions over approximately 6 weeks. The CTV should be larger for patients with HGAs for whom a margin beyond the entire lesion of at least 1.5 cm (or one vertebral body) would be more appropriate. The dose usually will be as for LGA because of the substantial risk of morbidity at higher doses but often chemotherapy will be given as well. While older studies99,100 and data from the Surveillance, Epidemiology, and End Results (SEER) registry106 and from the German cooperative group107 all suggest that 20% to 35% of children with high-grade gliomas will survive following such treatment, a recent study from St. Jude Children’s Research Hospital presents an even bleaker picture with a very high rate of both local progression and leptomeningeal spread and no long-term survivors among patients with GBM.105
 EPENDYMAL TUMORS
EPENDYMAL TUMORS
The following types are seen in children:
• Myxopapillary ependymoma (WHO grade I)
• Ependymoma (WHO grade II)
• Anaplastic ependymoma (WHO grade III)
Myxopapillary Ependymoma
Myxopapillary ependymomas are slowly growing lesions almost always located in the conus filum terminale region of the spinal cord. They are the most common spinal cord tumor in this location. They usually present with back pain. On imaging, myxopapillary ependymomas are well circumscribed and usually enhance brightly after contrast injection. Despite their low-grade histology, leptomeningeal spread is not uncommon even at diagnosis and all patients should have an MRI of the whole spine and brain as part of their initial workup.
Surgical resection is the treatment of choice. If the tumor is contained within the filum, complete resection may be possible after mobilization of the filum. Whether postoperative radiotherapy is necessary after complete resection is unclear. A significant proportion of tumors recur locally and/or with leptomeningeal metastases, but salvage with further surgery and/or with radiotherapy seems to be possible in most.108–110 If the tumor is in continuity with the conus, resection is more difficult and more likely to result in significant sequelae so that there frequently will be residual tumor. If the tumor is not resected en bloc or if there is macroscopic residual tumor, the risk of recurrence is high. Postoperative radiotherapy in this situation results in improved local control.101,111–113 The radiotherapy target volume is local (macroscopic disease plus a margin cephalad and caudad of 1.5 cm [or one vertebral body]) for the CTV and the dose, 50.4 Gy.113 Patients with leptomeningeal seeding at diagnosis or at relapse after surgery alone should be treated with curative intent with CSI followed by a boost to the primary site with a reasonable expectation of long-term tumor control.101,109,112,114
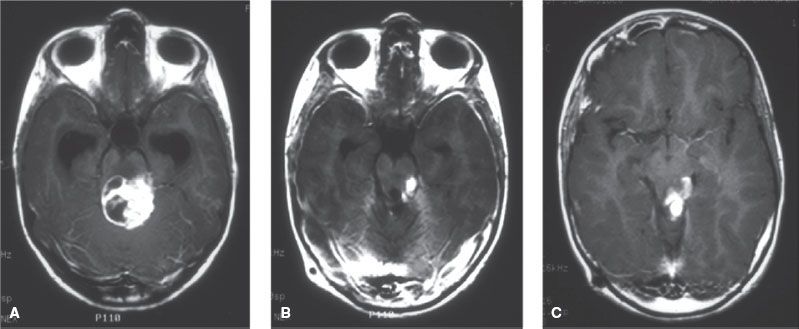
FIGURE 84.10. Typical appearance of an ependymoma that fills the fourth ventricle causing hydrocephalus and extends inferiorly below the foramen magnum over the dorsal aspect of the spinal cord to the level of C4.