Fig. 14.1
Brain areas crucial to mineralocorticoid-salt and renovascular hypertension
History
A century after Addison described patients with adrenal cortical destruction as having “asthenic” hearts, deoxycorticosterone, DOC, the first mineralocorticoid to be isolated and synthesized, and deoxycorticosterone acetate (DOCA) were used to normalize fluid and electrolyte balance and strengthened cardiodynamics in patients with Addison’s disease [14]. It was clear by then that overzealous replacement led to hypertension and renal damage in humans and experimental animals, particularly if paired with excess salt intake [2, 4, 5, 14]. Early studies demonstrated that sub-pressor doses of DOC augmented vascular responses to epinephrine and norepinephrine in healthy people on a normal salt diet, suggesting direct (vascular) and/or indirect (neural) effects on vascular tone led to the hypothesis that increased sympathetic drive was involved in essential hypertension [15].
Aldosterone, the primary endogenous mineralocorticoid, was isolated a few years later in 1952 [16–19], followed in 3 years by the description of the eponymous Conn’s Syndrome, or Primary Aldosteronism, associated with hypertension and hypokalemia, and eventual heart and kidney failure [20]. Notwithstanding early evidence of direct cardiovascular and neural effects of mineralocorticoids [15, 21, 22] and demonstration that DOCA produced hypertension in dialyzed animals after bilateral nephrectomy [23], retention of sodium and water by the kidneys became commonly accepted as primarily, if not exclusively, responsible for mineralocorticoid/salt excess hypertension, and the hypertension for pathological structural changes [2, 24, 25].
Mineralocorticoid Receptors in the Brain and Hypertension
Involvement of specific receptors for aldosterone in the neurohumoral control of cardiovascular and osmotic homeostasis was recognized by the 1960s [26, 27]. Autoradiographic studies demonstrated distinct receptors and binding characteristics for the two primary adrenal steroids, aldosterone and corticosterone, in the brain [28–30]. Among the areas of heaviest aldosterone binding are the hippocampus, circumventricular organ (CVO), some hypothalamic nuclei, and parts of the amygdala and brain stem. More recent immunohistochemical studies show that the MR is expressed in very specific neurons [31–34]. Ablation of specific hypothalamic and circumventricular areas, particularly an area anteroventral to the third ventricle, the AV3V area, prevented or attenuated the increase in blood pressure in renovascular and salt-sensitive forms of hypertension, including that of mineralocorticoid excess and some genetically hypertensive rat strains [35–41] (Fig. 14.1). Ablation of the AV3V area decreases sympathetic drive, baroreceptor sensitivity, and the systemic increases in vasopressin (VP) and serotonin produced by mineralocorticoid excess (reviewed in [42]). The AV3V area comprises the organum vasculosum of the lamina terminalis (OVLT), median preoptic nucleus, and afferents to the subfornical organ (SFO) and VP- and oxytocin-containing magnocellular neurons of the paraventricular nucleus (PVN). MR expression is dense in the OVLT, SFO and in the area postrema (AP), all CVO outside the blood–brain barrier, where changes in osmotic pressure and concentrations of sodium and a large variety of circulating hormones and cytokines in the blood and cerebrospinal fluid are detected. Information from CVOs is relayed to the hypothalamus and other brain areas, including those regulating sympathetic drive to the heart, vessels, and kidneys [43–46]. While AV3V lesions prevent activation of the central sympathetic nervous system (SNS) in response to mineralocorticoids and centrally angiotensin II and attenuate the pressor response to systemic nitric oxide inhibition [45], they do not attenuate the vasoconstrictor actions of neural- or adrenal-derived catecholamines [43].
Selective infusions of mineralocorticoid receptor (MR) agonists and antagonists in various dog and rat models of hypertension confirmed and extended information from the ablation studies (reviewed in [47, 48]). Inappropriate activation of CVO MR mediates hypertension through an increase in sympathetic drive to the peripheral cardiovascular and renal systems, as well as through the release of VP and decreased baroreceptor sensitivity [42, 49–53] (see Fig. 14.2). Activation of MR of the amygdala increases sodium appetite [54–56].
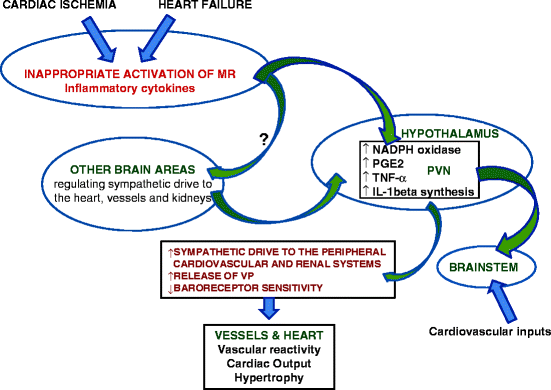
Fig. 14.2
Activation of MR in brain circumventricular organs during cardiac ischemia and heart failure paraventricular nucleus of hypothalamus (PVN), circumventricular organs (CVO), vasopressin (VP), see refs. [58, 60, 62] these are the primary and first to demonstrate the connection between heart damage, inflammation, the PVN and the SNS
Pivotal Role of the Central Sympathetic Nervous System
Patients with Primary Aldosteronism and renovascular hypertension have significantly greater sympathetic nerve activity than those with essential hypertension and non-hypertensive subjects and sympathetic nerve activity is normalized after surgical resolution of their disease [53, 57]. The persistent increase in sympathetic drive mediated by the activation of brain MR is responsible in part for the pathological structural remodeling and dysfunction following myocardial infarction in experimental animal models, even when circulating aldosterone levels are normal or low [58–63].
The central SNS is also activated by stress [64]. Cortisol, a major stress hormone, regulates the hypothalamic-pituitary-adrenal axis (HPA) through central glucocorticoid (GR) and mineralocorticoid receptors (MR). The activity of the HPA axis can be modulated by changes in function of the MR and GR [64–66].
MR, Inflammation, and Structural Changes in the Heart and Vessels
MR are involved and necessary in repair after tissue injury; however, prolonged and inappropriate activation leads to pathological remodeling and fibrosis. Inappropriate MR activation mediates the hypertrophy, inflammation, and fibrosis of the heart, vessels, and kidneys associated with Primary Aldosteronism [67–70], experimental mineralocorticoid/salt excess [6, 62, 71–75], and the hyperaldosteronism driven by elevated angiotensin II levels [72, 76–80]. These structural alterations and pathology can occur independently of significant hypertension [6, 48, 81, 82] and is preceded by inflammation in patients and animal models [6, 83–91]. The inappropriate activation of non-epithelial tissue MR also increases markers of oxidative stress, as well as diverse cellular mediators of inflammation in vitro and in vivo [92–97].
The progression of cardiac dysfunction to heart failure after experimental myocardial infarction is similar to that of mineralocorticoid excess and is mitigated by MR antagonists [58, 59, 98]. Inflammatory cytokines are involved in the communication between the injured heart and neurons in the PVN and AV3V area. Activation of MR in brain CVO during cardiac ischemia and heart failure leads to increased levels of neuronal activity, TNF-α and IL-1beta synthesis in the PVN, and TNF-α and IL-1beta levels in the blood and heart [59, 62, 99–104]. In addition, sympathetic afferent nerves from the injured heart signal to the brain to increase cytokine production [103] (Fig. 14.2).
Neurons of the AV3V area respond to IL-1beta, through a mechanism that includes the local synthesis of prostanoids, by exciting neurons of the medial preoptic area and PVN [105]. Circulating TNF-α and other proinflammatory cytokines induce cyclooxygenase (COX)-2 activity and synthesis of prostaglandin E2 by perivascular cells of vessels in the brain and thereby activate neurons in the PVN of hypothalamus, including corticotropin-releasing hormone (CRH) neurons. Treatment of a rat coronary ligation heart failure model with a MR antagonist significantly reduced cerebrospinal fluid prostaglandin E2, plasma norepinephrine, TNF-α, interleukin (IL)-1beta and IL-6, as well as hypothalamic COX-2 and CRH protein expression. The MR antagonist also decreased immunohistochemical staining in PVN neurons for TNF-α, interleukin (IL)-1beta, and CRH in the rat heart failure model. The effect of the MR antagonist was similar to that of etanercept and pentoxifylline, anti-cytokine drugs [106]. Along with amelioration of the inflammatory profile, left ventricular end-diastolic pressure and lower right ventricle/body weight and lung/body weight ratios were decreased; however, there was no improvement in left ventricular function of the MR antagonist-treated heart failure rats [103, 106]. These inflammatory responses are adaptive and necessary for the immediate response to injury and repair, presumably for the more common, less devastating insults to the myocardium [60]. Pretreatment and initiating MR antagonist treatment at the time of the coronary ligation decreased survival of the rats [60]. While the forebrain MR are crucial for repair and the short-term survival of cardiac ischemia induced by coronary ligation in the rat [60], their continued, excessive, and/or inappropriate activation exacerbates and accelerates the progression of heart failure due to inappropriate sympathetic nerve activity, volume retention, and inflammation that cannot be reversed with the systemic administration of antagonists of the renin-angiotensin system (RAS) [62].
Mineralocorticoid Receptor Biology
The MR is a member of the steroid nuclear receptor superfamily of ligand-activated transcription factors. Among these receptors, the MR shares the most homology with glucocorticoid receptors (GR). In addition to promoting gene transcription, MR also initiate rapid non-genomic effects through second messenger pathways [107–111] (reviewed by [109] and [112]). MR also appear to be activated in a steroid-independent fashion through transactivation by other signal transduction pathways, in particular that of angiotensin II [113].
The “Classical” genomic action of the MR is best understood (review [114]). Ligand-free MR is primarily located in the cytosol associated with chaperon and cell scaffolding proteins that alter the conformation of the molecule and anchor it in proximity to interacting proteins. Upon activation, MR are moved to the nucleus where they associate with the DNA as dimers [114–118]. MR form both homodimers and heterodimers with GR [119–121]. The effect of heterodimerization on transcriptional activity has not been completely elucidated, but is likely to have implications on glucocorticoid, as well as mineralocorticoid function [114, 122].
Like several other members of the steroid hormone transcription factor superfamily, notably the estrogen, androgen, and progesterone receptors, some MR are associated with the plasma membrane, probably in caveolae, and act through second messenger systems [109, 123–125]. Genomic or transcriptional effects mediated by MR take several hours to occur; rapid non-genomic MR effects are initiated within minutes upon MR activation and may also result in the transcription of effector genes with a latency of 3 or more hours. Many, if not most, effects mediated by the MR, including the action of mineralocorticoids in the brain to increase salt appetite, as well as inflammation and repair, are mediated by both genomic and more rapid non-genomic actions [126–128].
Mineralocorticoid-mediated stimulation of the epithelial Na+ channel and Na/K-ATPase pump were among the first MR-mediated intracellular effects studied, primarily in the nephron, colon, and toad bladder [129–131]. The increase in activity for both the ENaC and Na/K-ATPase are complex and multi-factorial, involving both genomic and non-genomic actions of MR leading to the net increase in channel activity and decrease in degradation [132–138]. Both ENaC and the Na/K-ATPase are instrumental in the central effects of aldosterone on blood pressure [139–142].
There is substantial evidence for an endogenous digitalis- or ouabain-like factor with an important role in the central control of the blood pressure [63, 142–148]. Endogenous ouabain is instrumental in the hypertension produced by the inappropriate activation of the MR during Na+ repletion or excess in clinical and experimental conditions [142, 149–151]. Despite more than 30 years of concerted effort, the mechanism for its synthesis in a mammal has not been elucidated nor have intermediates been isolated [152, 153].
Mineralocorticoid Receptor Has Two Physiological Ligands
The MR are bound and, depending on the context, can be activated by two classes of steroid, the mineralocorticoids, principally aldosterone, and the glucocorticoids, cortisol, and corticosterone. The isolated or recombinant MR, which has also been called the “type I corticosteroid receptor,” binds aldosterone, corticosterone, and cortisol with similar high affinity [154]. Circulating levels of aldosterone are regulated by Na+, K+, and angiotensin II in response to osmotic and hemodynamic need. Glucocorticoids are regulated by ACTH in a circadian manner and in response to energy needs and stress. As blood concentrations of glucocorticoids are normally 100–1,000 times greater than those of aldosterone, clearly circulating ligand concentrations are not the sole determinant of the extent of MR activation by aldosterone. MR splice variants so far described do not account for tissue-specific differences in MR ligand preference or activation specificity [155, 156]. MR specificity for aldosterone in epithelial mineralocorticoid target tissues is conferred extrinsically by the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) that converts cortisol and corticosterone to the inactive cortisone and 11-dehydrocorticosterone, allowing aldosterone access to the MR [157, 158]. 11β-HSD2 physically interacts with the MR within the endoplasmic reticulum membrane, thus the inactivation of cort and modulation of the relative levels of competing steroid occurs in very close proximity to the receptor [159–161]. Inactivating mutations of the 11β-HSD2 gene and pharmacological inhibition of the enzyme allow cortisol or corticosterone to activate renal tubular epithelial MR, producing Apparent Mineralocorticoid Excess characterized by Na+ and water retention, K+ and H+ excretion, hypertension, and suppresses the renin-angiotensin-aldosterone system (RAAS) [162]. While the consequences of activation of the MR by aldosterone or cort are the same for some functions, including Na+ retention in the kidney [163, 164], they are different for others, including many if not all of those in the CNS [165–171].
Another mechanism for 11β-HSD2 modulation of MR function has been proposed. MR in cells without 11β-HSD2 such as those in the hippocampus or heart are normally occupied by glucocorticoids and do not mediate inflammation or repair unless exposed to reactive oxygen species [89, 111, 172]. The hypothesis is that MR bound to glucocorticoids are inactive, including in cells that express 11β-HSD2. 11β-HSD2 requires NAD+ as cofactor for dehydrogenase activity and generates NADH [159]. Inhibition of 11β-HSD2 allows the accumulation of the reactive oxygen species NAD+, triggering the activation MR by glucocorticoids, for both specific functions normally attributed to aldosterone activation, as occurs in the renal tubule in AME, and inflammation and collagen formation, as occurs in the injured heart [173]. There are uncertainties about how 11β-HSD2 confers selectivity for aldosterone to the MR, yet it is clear that an understanding of the mechanisms that regulate glucocorticoid-activated MR activity is crucial to developing interventions into MR-mediated pathology, including the modulation of blood pressure and structural remodeling of the heart and vessels, by MR in the CNS, many of which do not appear to be associated with the 11β-HSD2 [174–176].
Autoradiography and functional tests demonstrate that MR in different parts of the brain have distinct preferences for either aldosterone or cort, with those in the hippocampus binding glucocorticoids and aldosterone in equal measure in adrenalectomized rats [29]. In vitro incubation of rat brain minces with tritiated precursors demonstrates the efficient conversion of corticosterone to the inactive steroid 11-dehydrocorticosterone [177, 178]. Moreover, intracerebroventricular infusions of three different 11beta-HSD inhibitors at doses too small to have an effect systemically produce hypertension. The central infusion of an MR antagonist abrogates the hypertension produced by the systemic administration of 11beta-HSD inhibitors, demonstrating a specific MR-driven response [47, 179, 180]. 11β-HSD2 mRNA was detected in the circumventricular nuclei by in situ hybridization [181] and both 11β-HSD2 and MR have been detected in the same neuron in select neurons of the NTS by immunohistochemistry [32–34].
As mentioned above, though some MR functions are initiated by either aldosterone or cort, others are ligand-specific [167, 171, 182]. The mechanisms for ligand-selectivity and functional specificity for MR in the brain are not adequately explained. In addition to the large differences between the circulating levels of aldosterone and glucocorticoids, there is the expression of the multiple drug resistance protein or p-glycoprotein that actively pumps specific compounds, including steroids out of cells, including those that form the blood brain barrier. p-glycoproteins are expressed in neurons, particularly those of the granular cell layer of the dentate gyrus, in addition to microvascular endothelial cells and astrocyte podocytes touching the microvessels of the brain [183]. In humans, transport by p-glycoprotein, i.e., exclusion from the brain, is significantly less for aldosterone than for cortisol, while corticosterone extrusion is minimal, suggesting that corticosterone, which circulates at 5–10% the level of cortisol in the human, is a significant ligand for the MR and GR in the human brain [184, 185]. Aldosterone concentrations in the rat brain are slightly lower than those in plasma, still 1–2 orders of magnitude levels of corticosterone, the major glucocorticoid in the rat [186]. Therefore for the Multiple Drug Resistant protein system to be a determinant of MR ligand specificity, it would have to function at the level of the individual neuron as it has been found in the hippocampus [183].
There is evidence for two other mechanisms that may increase local concentrations of aldosterone in or near aldosterone target cells: synthesis of aldosterone within the brain and the formation of an aldosterone derivative that is not a substrate for the p-gycoprotein and can activate the MR. Neurosteroids are those synthesized in the brain independently of the synthesis in the periphery [187, 188]. The requisite components for aldosterone synthesis, including StAR protein, required for cholesterol transport into the mitochondria, and the enzymes required for de novo aldosterone synthesis from cholesterol, including the aldo synthase enzyme unique to the formation of aldosterone, are expressed in rat and human brains [177, 178, 189–193]. Aldosterone synthase immunoreactivity is localized in neurons in several areas of the brain where MR expression is dense, including the cerebellum and hippocampus [194]. Aldosterone is produced in vitro from endogenous substrates, as well as tritiated deoxycorticosterone and corticosterone, by brain tissue from intact and adrenalectomized rats with similar efficiency [178, 190]. The aldosterone content of the brain reflects that of the plasma in adrenal-intact rats on diets of different salt content [193]. Extra-adrenal synthesis of aldosterone can only be appreciated by removal of the adrenals. In adrenalectomized rats the aldosterone content of the brain is very low, but consistently measurable and significantly higher than that of the plasma, which is usually below the limits of detection [193]. These data suggest that aldosterone synthesis does occur in the brain, but that normally most of the brain aldosterone is derived from the adrenal gland. Inappropriate aldosterone production within the brain appears to be involved in the pathogenesis of hypertension in the Dahl Salt Sensitive rat. Salt-induced hypertension in the Dahl SS is prevented by the central infusion of a MR antagonist, as well as AV3V ablation, implicating MR in the CVO in the hypertension, even though circulating aldosterone in these rats is suppressed by the high salt diet [195]. The salt-sensitive hypertension in the Dahl SS rat is also mitigated by the central infusion of inhibitors of steroidogenic enzymes, including one specific for aldosterone synthase, implicating aldosterone synthesis within the brain in this model [176, 177, 196]. Transgenic rats expressing the aldosterone synthase enzyme driven by the Synapsin 1 promoter which targets neurons have hypertension. Like the Dahl SS, circulating aldosterone levels are normal (unpublished data).
Aldosterone production by the adrenal is tightly regulated primarily by dietary sodium and angiotensin II; however, information about the regulation of the brain RAS, including the AT1 receptor, by sodium status in areas associated with hemodynamic homeostasis has been inconsistent [197–200]. However, inhibition of the central, but not systemic RAAS, decreases sympathetic drive and activation of the peripheral RAAS and increases tissue and circulating inflammatory cytokines produced by coronary ligation, suggesting that synthesis of aldosterone within the brain is regulated by the local RAAS [58, 59].
Aldosterone, cortisol, corticosterone, testosterone, and estradiol circulate as esters of fatty acids that are more hydrophobic than the parent compound, thus concentrate in tissues, particularly the brain and fat [201, 202], and regulation of the synthesis of esters may be a mechanism to control tissue concentration of these steroids [203, 204]. Both estrogen and aldosterone esters are potent activators of their respective receptors; some aldosterone esters have greater biological activity than aldosterone [205–209]. Aldosterone esters are hydrolysed to aldosterone very rapidly in blood, plasma, and serum and are not differentiated from aldosterone by common methods of measurement of aldosterone [209]. We confirmed studies done by the late Mary Lockett demonstrating the existence of significant amounts of an aldosterone ester in the heart and brain and found that aldosterone-18-monoacetate and -21-mono-oleate are more potent as hypertensinogens when infused intracerebroventricularly than the parent compound aldosterone [209]. As the enzymes responsible for steroid esterification are specific to the steroid [204], one could speculate that the local regulation of aldosterone ester formation may be a mechanism to exert aldosterone selectivity for the MR.
An area of study with great potential for finding mechanisms for MR ligand-selectivity and ligand functional specificity in different cells is that of the differential distribution of proteins which interact with steroid receptors [114]. Cell type-specific chaperone proteins may alter the conformation, thus binding affinity for aldosterone of the MR compared to the glucocorticoids in some tissues. Upon binding to a ligand, the MR sheds the chaperone proteins and dimerizes either as a homo or heterodimer with the GR in the nucleus, before or upon binding to hormone response elements in the promoter regions of specific genes, initiating the expression of these genes by recruiting requisite transcriptional machinery [120, 210–214]
Review [114]. MR and glucocorticoid receptors (GR) are co-expressed in many cells of the brain, including cerebellar purkinje and hippocampal pyramidal cells [215], where heterodimerization of MR and GR appears to be important in the transcriptional regulation of glucocorticoid-responsive genes in the brain [122] [216]. It is probable that GR and MR heterodimerization figures in the control of blood pressure and neurohumoral control of peripheral inflammation. Transcription efficiency is modulated by co-activators or co-repressors, proteins that bind activated receptors. To date, there are no cofactors or hormone response elements known to be specific for the MR; however, Steroid Receptor Co-activator protein family members have distinct anatomical distribution in the brain, with expression of SRC-1a mRNA prominent in the circumventricular areas of the brain associated with blood pressure homeostasis, as well as mineralocorticoid hypertension [114, 217–219].
In conclusion, mineralocorticoid receptors in specific neurons of the brain, primarily those of the CVO, hypothalamus, and amygdala mediate hemodynamic control, as well as modulate inflammation and repair in peripheral target organs. However, most of what is known is about their role in pathological conditions; we know very little about the function of MR in the brains of healthy individuals. MR antagonists greatly ameliorate and slow the progress of heart and renal failure and their therapeutic use continues to increase, despite the fact that the preponderance of MR in the brain has functions unrelated to cardiovascular homeostasis or inflammation. More must be learned about the basic biology and function of the MR in the various areas of the brain to guide the development of targeted therapeutic agents.
Acknowledgments
This work was supported by Merit Review Medical Research funds from the Department of Veterans Affairs and NIH grants HL27255, HL27737 and HL75321. We are grateful to Dr. Eugen Melcescu who helped with the illustrations.
There are no conflicts of interest.
References
1.
Steiger M, Reichstein T. Desoxy-corticosterone (21-oxy-progesterone). Aus d-3-oxo-etiocholensäure. Helv Chim Acta. 1937;20:1164.
2.
Kuhlman D, Ragan C, Ferrebee JW, Atchley DW, Loeb RF. Toxic effects of deoxycorticosterone esters in dogs. Science. 1939;90:496–7.
3.
Rodbard S, Freed SC. The effect of desoxycorticosterone acetate on the blood pressure of the dog. Endocrinology. 1942;30:365–8.
4.
Luft R, Sjogren B. The effect of desoxycorticosterone acetate and sodium chloride on blood pressure and renal function. Acta Endocrinol (Copenh). 1949;3(1):56–70.
5.
Knowlton AI, Loeb EN, et al. Desoxycorticosterone acetate studies on the reversibility of its effect on blood pressure and renal damage in rats. Endocrinology. 1949;45(4):435–45.
6.
Gomez-Sanchez EP. Brain mineralocorticoid receptors: orchestrators of hypertension and end-organ disease. Curr Opin Nephrol Hypertens. 2004;13(2):191–6.
7.
Pitt B, Zannad F, Cody R, Castaigne APA, Palensky J, Wittes J. The effect of spironolactone on mobidity and mortality in patients with severe heart failure. Randomized Aldactone evaluation study investigators. N Engl J Med. 1999;341:709–17.
8.
Gomez-Sanchez CE, Rossi GP, Fallo F, Mannelli M. Progress in primary aldosteronism: present challenges and perspectives. Horm Metab Res. 2010;42(6):374–81.
Related posts:
 Congenital Adrenal Hyperplasia
Insulin Resistance and Hypertension
Congenital Adrenal Hyperplasia
Insulin Resistance and Hypertension
 Primary Hyperparathyroidism and Hypertension
Primary Hyperparathyroidism and Hypertension
 Testosterone Deficiency or Male Hypogonadism
Testosterone Deficiency or Male Hypogonadism
 Primary Generalized Familial and Sporadic Glucocorticoid Resistance (Chrousos Syndrome) and Hypersensitivity
Primary Generalized Familial and Sporadic Glucocorticoid Resistance (Chrousos Syndrome) and Hypersensitivity
 Primary Aldosteronism: Progress in Diagnosis, Therapy, and Genetics
Primary Aldosteronism: Progress in Diagnosis, Therapy, and Genetics
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


