INTRODUCTION
The efficacy of allogeneic hematopoietic cell transplantation (HCT) in hematologic malignancies can in large part be attributed to a graft-versus-tumor (GVT) effect, by which the donor immune system achieves immunologic control of the tumor. As such, it is the prototype of cellular therapy. The human leukocyte antigen (HLA) system is fundamental to transplant biology. The HLAs are highly polymorphic proteins that have a key role in antigen presentation and immunoregulation. Class I HLAs are expressed on the surfaces of all nucleated cells; class II are expressed on specialized antigen-presenting cells (APCs), such as macrophages, dendritic cells, and B cells.
Peptides derived from microbes are presented on class I HLAs to CD8+ T cells and result in immunologic destruction of infected cells; class II HLAs are recognized by CD4+ T cells. T-cell activation requires costimulatory signals from the APC, specifically CD80/86 binding to CD28 or LFA-3 binding to CD2 (1). Absence of a costimulatory signal results in T-cell anergy, which is a key mechanism of peripheral immune tolerance to self-antigen in normal immunoregulation. Early posttransplantation, there is a “cytokine storm”; release of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), is induced by tissue damage from the conditioning regimen, activating the host innate immune system (Fig. 16-1). Donor T cells interact with host APCs and recognize foreign peptides; helper T cells produce further cytokines, especially IL-2, and prime host APCs via CD40:CD40L interaction. Differentiation of naïve donor T cells into effector cells subsequently occurs, resulting in immunologic attack on host tissues and the potential development of graft-versus-host disease (GVHD) (2). Increasing numbers of HLA mismatches are associated with higher incidence of GVHD and transplant-related mortality (TRM) (3). However, even in a fully HLA matched HCT, GVHD still occurs due to donor T cells directed against minor histocompatibility antigens (MiHAs), polymorphic peptides displayed on host HLA molecules.
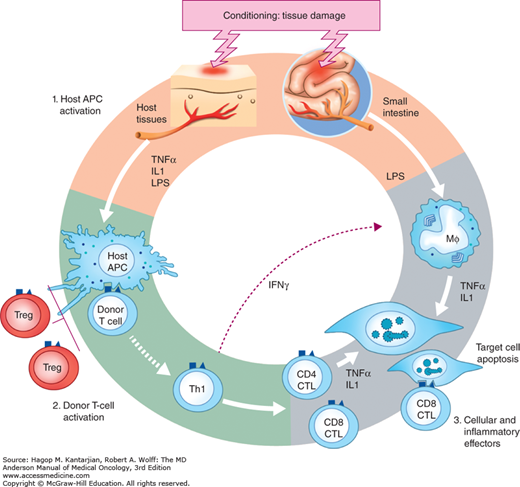
FIGURE 16-1
Pathogenesis of GVHD. In phase I, chemotherapy or radiotherapy as part of transplant conditioning causes host tissue damage and release of inflammatory cytokines such as TNF-α, IL-1, and IL-6, with resulting priming of host antigen-presenting cells (APCs). In phase II, host APCs activate mature donor cells, which subsequently proliferate and differentiate; release of additional effector molecules, such as TNF-α and IL-1, mediates further tissue damage. Lipopolysaccharide (LPS) that has leaked through damaged intestinal mucosa triggers additional TNF-α production. The TNF-α can damage tissue directly by inducing necrosis and apoptosis in the skin and gastrointestinal tract through either TNF receptors or the Fas pathway. Tumor necrosis factor alpha plays a direct role in intestinal GVHD damage, which further amplifies damage in the skin, liver, and lung in a “cytokine storm.” The process culminates in death of host cells through CD8-positive cytotoxic T-cell-mediated apoptosis. [Reproduced with permission from Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550-1561].
There are three key barriers to successful HCT: GVHD, infectious complications, and relapse due to failure of immunologic control of the underlying disease. Herein, we review these issues in greater detail with an emphasis on recent cellular therapeutic approaches to address these complications.
ENHANCING GRAFT-VERSUS-TUMOR EFFECT TO OVERCOME TUMOR ESCAPE
The GVT effect occurs due to a predominantly T-cell-mediated immunologic attack on tumor cells. In HLA-identical transplants, GVT effect is mediated by naïve T cells; for development of effector function, these must first be primed by host APCs. This requires the following: presentation of MiHAs or tumor-specific antigens on HLA; appropriate costimulatory molecules, including CD28, OX40, CD40L, and 41BB; and an appropriate “third signal,” provided by IL-12, interferon gamma (IFN-γ) or adjuvant (4). Restraining influences limiting the degree of immune activation are present to protect the host from an excessive immune response and include expression of CTLA4 (which competes with CD28 for binding to CD80/86) and programmed cell death protein 1 (PD1) and its interactions with its lig and PDL1, which limit T-cell activation and expansion during normal, pathogen-directed immune responses.
Soon after transplant, there is activation and expansion of MiHA-reactive T cells, followed by a decline, similar to that seen in pathogen-directed immune reactions. This may in part relate to the development of peripheral tolerance/anergy and also to replacement of host hematopoiesis, with resulting loss of host APCs. For tumor-associated antigen presentation to continue, there must be cross presentation on donor APCs. In addition, the initial alloresponse to MiHAs results in recruitment of T cells targeting either tumor-associated antigens or nonpolymorphic genes, which are either overexpressed or aberrantly expressed by the tumor (4).
Some tumor-associated antigens of importance include pathogenesis related protein (PR1), an epitope shared by proteinase-3 and elastase proteins, which is expressed in normal neutrophils and overexpressed in myeloid leukemias; and PR1-specific CD8+ T-cell responses, which can be detected in a range of myeloid and nonmyeloid malignancies post-HCT and correlate with outcome in chronic myelogenous leukemia (CML) (5). CD8+ T-cell responses to WT1, which is frequently overexpressed in acute myelogenous leukemia (AML), can be induced with vaccination strategies in both the autologous and allogeneic settings (6,7,8). Clinical responses from tumor vaccines have thus far been suboptimal.
Natural killer (NK) cells can also mediate antitumor effects; this is discussed in further detail in this chapter.
Tumors utilize numerous mechanisms to escape immunological destruction, including the following:
Induction of regulatory T cells
Production of inhibitory cytokines
Downregulation of costimulatory molecules (9) and HLA I (10)
Induction of coinhibitory molecules
Induction of myeloid-derived suppressor cells in the microenvironment that inhibit immune responses through multiple mechanisms.
Invasion of immunologically privileged sites (11).
Cellular therapeutic approaches are designed with the intent to abrogate these escape mechanisms.
Donor lymphocyte infusion (DLI) may induce remissions in patients with molecular or overt relapse of their malignancy and can reverse CD8+ T-cell exhaustion. (12). However, the likelihood of success varies significantly according to the underlying disease. Chronic myelogenous leukemia is most sensitive; follicular lymphoma (FL) and Hodgkin lymphoma (HL) are also highly responsive (13,14). Responses to DLI in AML or myelodysplastic syndrome (MDS) are less frequent, and durability is often poor. Acute lymphoblastic leukemia (ALL) is the least responsive to DLI. Reported response rates are 60% to 73% in CML, 15% to 29% in AML, and 0% to 18% in ALL (15,16).
Graft-versus-host disease occurs in 40% to 60% of patients with HCT (15,16) and is more likely to occur in unrelated donor recipients (13). Lympho-depleting chemotherapy given prior to DLI enhances alloreactive T-cell proliferation, potentiating the GVT effect but increasing GVHD (17). Approaches to reduce the GVHD incidence include the following:
Reducing T-cell dose. A dose-response relationship exists for both GVT and GVHD effects. In CML, no increased response is seen with cell doses greater than 4.5 × 108 CD3+ cells/kg. In AML, response rates plateau beyond 1.5 × 108/kg; higher doses increase GVHD. Gradual dose escalation schedules have been successfully utilized in relapses of indolent diseases. Follicular lymphoma appears highly sensitive to DLI after T-cell-depleted HCT, responding to low-dose DLI at doses from 1 to 10 × 106/kg with less than 20% incidence of clinically significant acute GVHD (aGVHD) and chronic GVHD (cGVHD) (14). A similar strategy has shown success in HL, mainly in the setting of low-volume disease detected on surveillance positron emission tomographic (PET) imaging (18). More sensitive surveillance techniques, such as deep sequencing, to detect low-volume disease may allow earlier institution of DLI and maximize efficacy; utilization of these techniques remains experimental.
Transduction of donor lymphocytes with a suicide gene (19).
Selection of T cells to target tumor-associated antigens/antigens with restricted or differential expression (analogous to the use of viral-specific T cells [VSTs]). Infusion of MiHA-specific T cells was effective at eradicating tumor in mouse models (20). In addition, tumor-infiltrating lymphocytes (TILs) recognizing tumor-associated antigens have been successfully used for selected metastatic solid tumors (21). However, the technology to isolate TILs remains restricted to a few specialized centers and is yet to be applied in large-scale clinical studies.
If there is insufficient residual donor hematopoiesis prior to DLI, eradication of host hematopoiesis by the infused lymphocytes can result in marrow aplasia; chimerism studies should therefore be performed prior to DLI to ensure adequate donor hematopoiesis (22).
Responses to DLI may not be seen for up to 2 months (11). In indolent diseases (eg, FL), this may not be problematic, but in overt relapse of aggressive diseases (eg, AML), chemotherapy may be required first to achieve disease control (23).
The delayed onset of action of DLI has led to the use of preemptive DLI to prevent relapse in high-risk patients. Mixed donor/recipient chimerism within the T-cell lineage is frequently seen in T-cell-depleted transplants and is associated with higher rates of relapse in CML (24), FL (14), and HL (18), likely due to development of bidirectional tolerance with resulting tumor escape from immunologic control. Donor lymphocyte infusion can induce full donor chimerism in both FL and HL, and subsequent relapse rates are low; no formal comparison to similar groups not receiving DLI has been performed.
The topic of bi-specific T-cell-engaging antibodies has been discussed in detail elsewhere. Bi-specific T-cell-engaging antibodies are single-chain antibodies that engage T cells via CD3 and direct them to an antigenic target present on tumor cells, typically CD19, resulting in T-cell redistribution, activation, expansion, and perforin-mediated killing of target cells (25). Blinatumomab is highly efficacious in ALL in the setting of persistent MRD (25), overt relapse, or refractory disease (26).
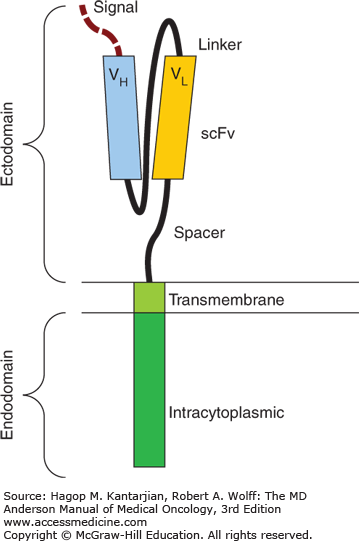
The ideal cellular therapy for a malignant disease should expand and persist in vivo and selectively target cancer cells. This can be achieved by modifying autologous T cells with a chimeric antigen receptor (CAR). The CAR consists of a single-chain monoclonal antibody (scFv) targeted to a tumor-associated antigen, which is thus recognized in a major histocompatibility complex (MHC)–independent fashion (unlike unmodified T-cell-mediated GVT effect); the scFv is coupled via an extracellular hinge domain and transmembrane domain to an intracellular signaling domain, typically the CD3ζ chain (Fig. 16-2) (27). Autologous T cells are collected from peripheral blood (PB) via a steady-state blood draw or apheresis procedure and transduced with the CAR construct via a lentiviral or retroviral vector or using electroporation and a transposon/transposase system (28). Cells are cultured and expanded ex vivo using either CD3/28 beads (29) or artificial APCs (30) and specific cytokines prior to infusion. The persistence and clinical activity of CAR T cells (CAR-T) in vivo can be enhanced by the addition of a costimulatory molecule to the CAR construct (31), usually CD28 (31,32). An initial report in a patient with highly refractory CLL treated with an anti-CD19 CAR-T utilizing CD137 (4-1BB) as the costimulatory domain generated great excitement (33): Infused T cells expanded more than 3 log, the patient developed a cytokine-release syndrome (CRS) and tumor lysis syndrome (TLS), and achieved complete remission (CR); long-term persistence of CAR-T and persistent normal B-cell aplasia (a predictable, on-target effect when targeting CD19) were demonstrated.
FIGURE 16-2
Schematic of basic chimeric antigen receptor (CAR) construct. The CAR consists of a single-chain monoclonal antibody (scFv) targeted to a tumor-associated antigen, and the scFv is then coupled via an extracellular hinge domain and transmembrane domain to an intracytoplasmic signaling domain, typically the CD3ζ chain.
Subsequent results in CLL have been heterogeneous; updated results from the University of Pennsylvania showed that 5 of 24 patients treated achieved durable CRs, 7 had partial responses (PRs), and 12 had no response (34). The variables underlying response to treatment are not well understood, but in vivo CAR-T expansion is a prerequisite and appears to be more important than the dose of infused cells (30). In ALL, results have been particularly impressive, with a CR rate of 86% in children treated for relapsed/refractory disease; patients were MRD negative even when tested with highly sensitive deep-sequencing techniques (34). Long-term survival rates are not yet known, particularly as in many cases the treatment has been used as a “bridge to transplant.” At least two patients with ALL have relapsed with CD19-negative disease (34). Similarly impressive CR rates have been reported by the groups at the National Institutes of Health (35) and Memorial Sloane Kettering Cancer Center (36). All three groups reported a similar toxicity profile (see Table 16-1). It is unclear whether this treatment can replace HCT in a proportion of patients; long-term survival outcomes in patients ineligible for HCT will be important in answering this question. The CAR-T targeting CD19 have now been used in a range of B-cell malignancies, with responses seen in both aggressive and indolent lymphomas, CLL, and ALL (Table 16-1).
| Reference | Cancer | CAR Endodomains | Number of Patients | Clinical Outcome | Toxicities |
|---|---|---|---|---|---|
| Kochenderfer et al, 2014 (91) | CLL, DLBCL, NHL, PMBCL, SMZL | CD28 & CD3ζ | 15 | 8 CR, 4 PR, 1 SD, 2 NE | Fever, hypotension, renal failure, confusion, aphasia |
| Maude et al, 2014 (35) | ALL | CD28 & CD3ζ | 30 | 90% CR (15 prior HCT); 67% EFS, 78% OS at 6 mo. | SIRS 27%, B-cell aplasia 73% |
| Lee et al, 2014 (36) | ALL | CD28 & CD3ζ | 21 | 70% CR | 33% severe SIRS |
| Davila et al, 2014 (35) | ALL | CD28 & CD3ζ | 16 | 88% CR | 43% severe SIRS |
| Kochenderfer et al, 2013 (92) | CLL, Lymphoma | CD28 & CD3ζ | 10 | 1 CR, 1 PR, 2 PD, 6 SD | Fever, SIRS, TLS |
| Brentjens et al, 2013 (93) | ALL | CD28 & CD3ζ | 5 | 5 CR | SIRS |
| Grupp et al, 2013 (94) | ALL | 4-1BB & CD3ζ | 2 | 2 CR | SIRS, central nervous system toxicity |
| Kochenderfer et al, 2012 (38) | CLL, Lymphoma | CD28 & CD3ζ | 8 | 1 CR, 5 PR, 1 SD, 1 died (influenza) | Mild SIRS |
| Brentjens et al, 2011 (32) | CLL, ALL | CD28 & CD3ζ | 8 | 1 PR, 2 SD, 3 NR, 1 PD, 1 died (sepsis-like disease) | Fever, death |
| Savoldo et al, 2011 (31) | NHL | CD28 & CD3ζ versus CD3ζ | 6 | 2 SD, 4 NR | None |
| Porter et al, 2011 (33) | CLL | 4-1BB & CD3ζ | 1 | CR | TLS, SIRS |
| Kalos et al, 2011 (95) | CLL | 4-1BB & CD3ζ | 3 | 2 CR, 1 PR | Fever, rigors, dyspnea, cardiac dysfunction, febrile syndrome, hypotension |
| Kochenderfer et al, 2010 (96) | Lymphoma | CD28 & CD3ζ | 1 | PR | None |
| Jensen et al, 2010 (27) | Lymphoma | CD3ζ | 2 | 2 NR | None |
While this therapy shows great promise, many aspects require optimization. Due to the heterogeneity in technique for CAR-T production, it is difficult to compare across trials to determine optimal manufacturing methods. The controversies are numerous: First, while it has been shown that addition of a costimulatory domain to the CAR enhances expansion and persistence (31), it is unclear whether CD28 or 4-1BB, or both, is superior. Second, lympho-depleting chemotherapy may enhance CAR-T expansion and persistence (32), but the optimal drugs and schedule are unknown. Third, the epitope targeted by the antibody fragment is likely important, in terms of both the spatial location of epitope binding and binding affinity. Fourth, the hinge and transmembrane domains are important in determining interaction with antigen and formation of the immunologic synapse, but little is known about optimizing this aspect of the CAR design. Fifth, the method of T-cell transduction (viral vs transposon) may be important in determining efficacy. Sixth, the ex vivo culture technique and duration (eg, CD3/28 beads vs artificial APCs and which supplemental cytokines to provide) may be important; for example, culture after transduction using the Sleeping Beauty system is relatively prolonged, which is problematic in kinetically active diseases such as ALL (37). The dose and composition (unselected vs specific ratios of CD4/8 cells) of T-cell product infused are also variables requiring consideration. Finally, the bulk of tumor present at the time of CAR-T infusion may potentially affect the in vivo proliferation of the infused CAR-T, and the ideal time of infusion (eg, in an MRD state vs overt relapse) has not been elucidated.
All responding patients have had some degree of CRS and have developed B-cell aplasia (38). Cytokine-release syndrome is characterized by fever with variable systemic symptoms, including hypotension, and high levels of inflammatory cytokines, of which IL-6 appears particularly important (37). Macrophage activation syndrome (MAS) may accompany CRS. Major neurological symptoms, including seizures, have occurred. The mechanism of neurological events is unclear; it may be cytokine mediated, associated with MAS, or due to direct CAR-T infiltration. Unexpectedly, CD19 CAR-T have been found in the cerebrospinal fluid of some patients without central nervous system disease (37). Cytokine-release syndrome can be managed with the anti-IL-6 antibody tocilizumab, with prompt responses in the majority of patients (37). Corticosteroids, while potentially efficacious in managing CRS/MAS, are toxic to the infused cells and may limit efficacy.
Unanticipated on-target toxicity may occur; for example, a toxic death occurred in a patient treated with a CART-T directed against Human Epidermal Growth Factor Receptor 2, or ErbB2, due to unanticipated low-level pulmonary epithelial expression (39). The inclusion of a suicide gene within the CAR, such as inducible caspase 9 (iCaspase9), which could be triggered in the event of severe toxicity, would provide an added safety measure.
Most human trials to date have focused on CD19 as a target in B-cell diseases, but a number of novel targets show potential in different diseases.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


