Cellular Signaling Pathways
8.1 INTRODUCTION
The ability of cells to receive and respond to extracellular signals is a critical process in the embryonic development of multicellular organisms as well as for the maintenance and survival of mature tissues in the adult. Changes in the physical or chemical environment of the cell can result in modifications of cell metabolism, morphology, movement, or proliferation. These responses are brought about by elaborate networks of intracellular signals transmitted by changes in protein phosphorylation and enzymatic activity, localization and the formation of protein–protein complexes. Cellular responses are triggered by the recognition of extracellular signals at the cell surface, resulting in the activation of linked cytoplasmic and nuclear biochemical cascades. These signal transduction pathways control cellular processes that range from the generalized control of cell proliferation and survival to specialized functions such as the immune response and angiogenesis. When dysregulated, signaling pathways involved in normal growth, adhesion and development contribute to malignant transformation in human cells. This knowledge has led to the development of new cancer therapeutics that specifically target aberrant signal transduction pathways. This chapter explores how signal transduction pathways are organized and highlights ongoing drug discovery efforts to target these pathways.
8.2 GROWTH FACTOR SIGNALING PATHWAYS
8.2.1 Extracellular Growth Factors and Receptor Tyrosine Kinases
In multicellular organisms, cell regulation is controlled by secreted polypeptide molecules called growth factors or cytokines, by antigen stimulation of immune cells, or by cell contact with neighboring cells and surrounding extracellular matrix. Our most detailed understanding of signal transduction pathways comes from studies of soluble growth factors and their interaction with complementary growth factor receptors expressed on responsive cells. The interaction between growth factors and receptors on the cell surface leads to the modification of intracellular biochemical signaling pathways that control cellular responses, especially cell proliferation. Cellular regulation also occurs through direct cell to cell contact or cell contact with its surrounding extracellular matrix (as discussed in Chap. 10, Sec. 10.2).
Growth factors were first identified in cell culture medium as necessary to sustain mammalian cell survival and proliferation. One of the characteristics of malignant transformation was found to be relative independence from the action of external growth factors. Many polypeptide growth factors have been identified with diverse functions in normal embryonic development and tissue homeostasis but only a few factors are associated with the process of malignant transformation.
Polypeptide growth factors influence cell processes, such as growth, proliferation, differentiation, survival, and metabolism, via their interaction with specific transmembrane receptor protein tyrosine kinases (RPTKs). Most are small monomeric (ie, single-chain) polypeptides, such as the epidermal growth factor (EGF) and members of the fibroblast growth factor (FGF) family. There are also dimeric polypeptide growth factors (ie, those containing 2 chains of amino acids), such as platelet-derived growth factor (PDGF). In addition to being freely diffusible, growth factors can also reside in spatially restricted domains within an organism, either through binding to components in the extracellular matrix or because they are produced as membrane anchored molecules that reside on the surface of the producing cells. Figure 8–1 summarizes selected growth factors and their cognate receptors.
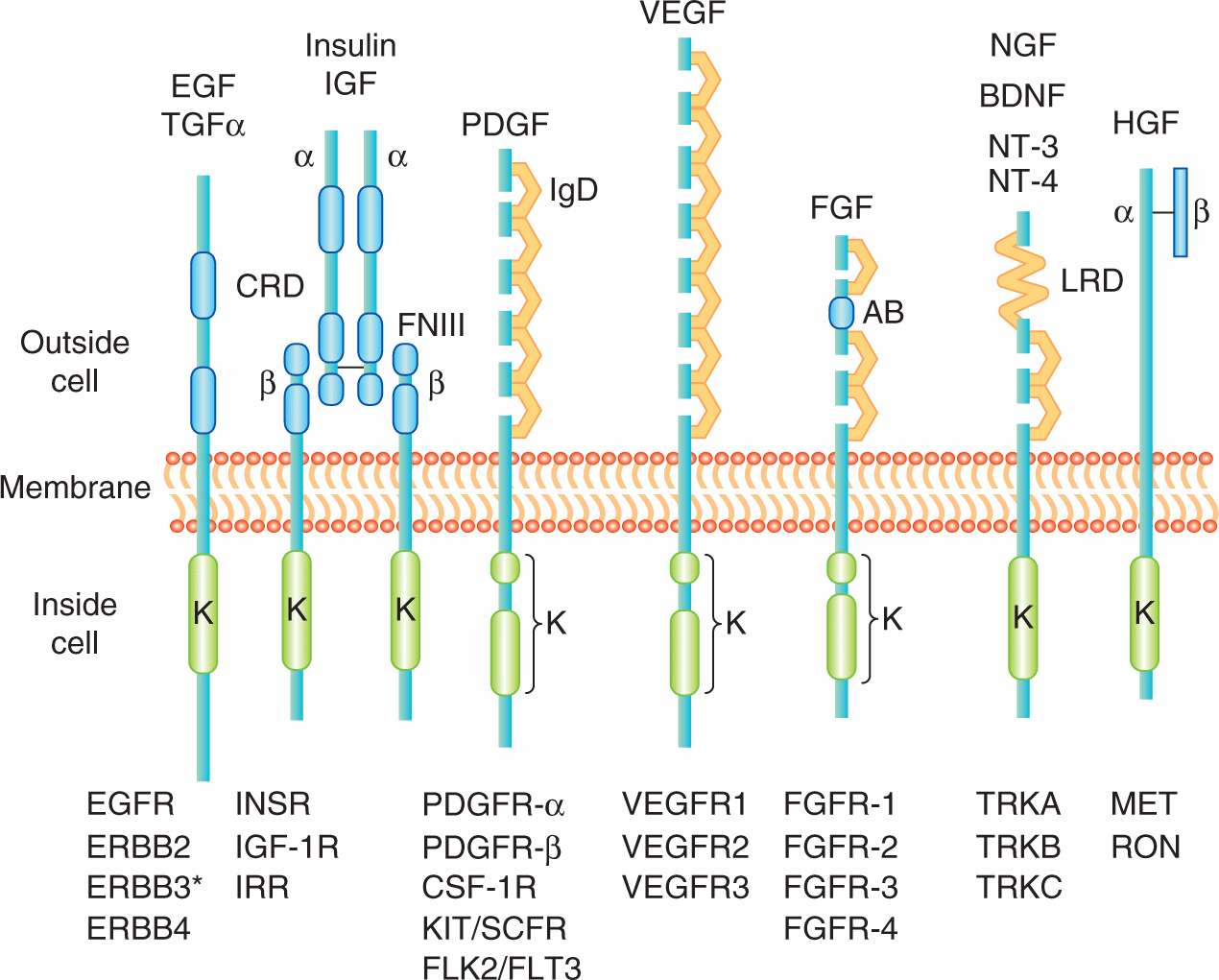
FIGURE 8–1 The receptor protein tyrosine kinases. Representative molecules from selected RPTK families of receptors (“R”) are shown below and representative ligands above. EGF, epidermal growth factor; FGF, fibroblast growth factor; HGF, hepatocyte growth factor; NGF, nerve growth factor; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor. All members have a conserved intracellular kinase domain (K). Some of the common structural elements found in the extracellular ligand binding domain include the CRD (cysteine-rich domain), FNIII (fibronectin type III repeats), IgD (immunoglobulin-like domain), AB (acid-rich box), and LRD (leucine-rich domain); TGF-α, transforming growth factor-alpha; IGF, insulin-like growth factor; BDNF, brain derived neurotrophic factor; NT-3, neurotrophin 3; NT-4, neurotrophin 4.
Receptors for growth factors are membrane spanning cell surface molecules that share the ability to phosphorylate themselves and other cytoplasmic proteins, thereby activating a signaling cascade. Most growth factor receptors are protein tyrosine kinases that specifically phosphorylate the amino acid tyrosine in protein substrates and can be subdivided into 20 different families based on distinct structural components. The main distinguishing feature amongst RPTK subgroups resides in the extracellular growth factor-binding domain at the amino terminus. Usually several hundred amino acids in length, these extracellular domains can be grouped by sequence homology, or by the presence of sequence motifs also found in other functionally unrelated molecules such EGF repeats, immunoglobulin repeats or fibronectin type III repeats (see Fig. 8–1). The extracellular domains of RPTKs are also commonly posttranslationally modified by glycosylation.
The extracellular domain is connected to the intracellular (cytoplasmic) domain by a short, single, hydrophobic helix transmembrane component. The cytoplasmic domain is comprised of regulatory sequences and a conserved kinase domain, which catalyzes the transfer of a phosphate group from adenosine triphosphate (ATP) onto a protein substrate. In RPTKs, the core catalytic domain is typically about 260 residues in length and is as much as 90% identical between members of this protein kinase family. The amino acids flanking the kinase domain and adjacent to the plasma membrane (juxtamembrane region) of growth factor receptors frequently contain sites of tyrosine phosphorylation and these regions often have important roles in both signal transmission and in regulation of catalytic activity. Such regulatory sequences can also reside within the catalytic domain of receptor kinases. Members of the PDGFR and vascular endothelial growth factor (VEGFR) families of receptors are distinguished by possessing a split kinase domain in which important autophosphorylation sites are present on a kinase insert within the catalytic domain (van der Geer et al, 1994).
Binding of the growth factor or ligand induces conformational changes in the extracellular domain of the receptor that facilitate dimerization (ie, joining together) or clustering of receptor tyrosine kinases (Fig. 8–2). Some ligands, such as PDGF, are themselves dimeric forms of a single subunit and naturally induce a symmetric ligand/receptor dimer. Structural studies have revealed how other ligands that exist as monomers, such as EGF, induce receptor dimerization through receptor-receptor interactions. These studies revealed that binding of EGF to the epidermal growth factor receptor (EGFR) (ERBB1) induces a conformation change that exposes a dimerization loop that mediates association of neighboring, ligand occupied receptors (Schlessinger, 2002). Similar dimerization loops are found in the other members of the EGFR family (ERBB2, ERBB3, and ERBB4) allowing the formation of heterodimers between different members of the ERBB family.
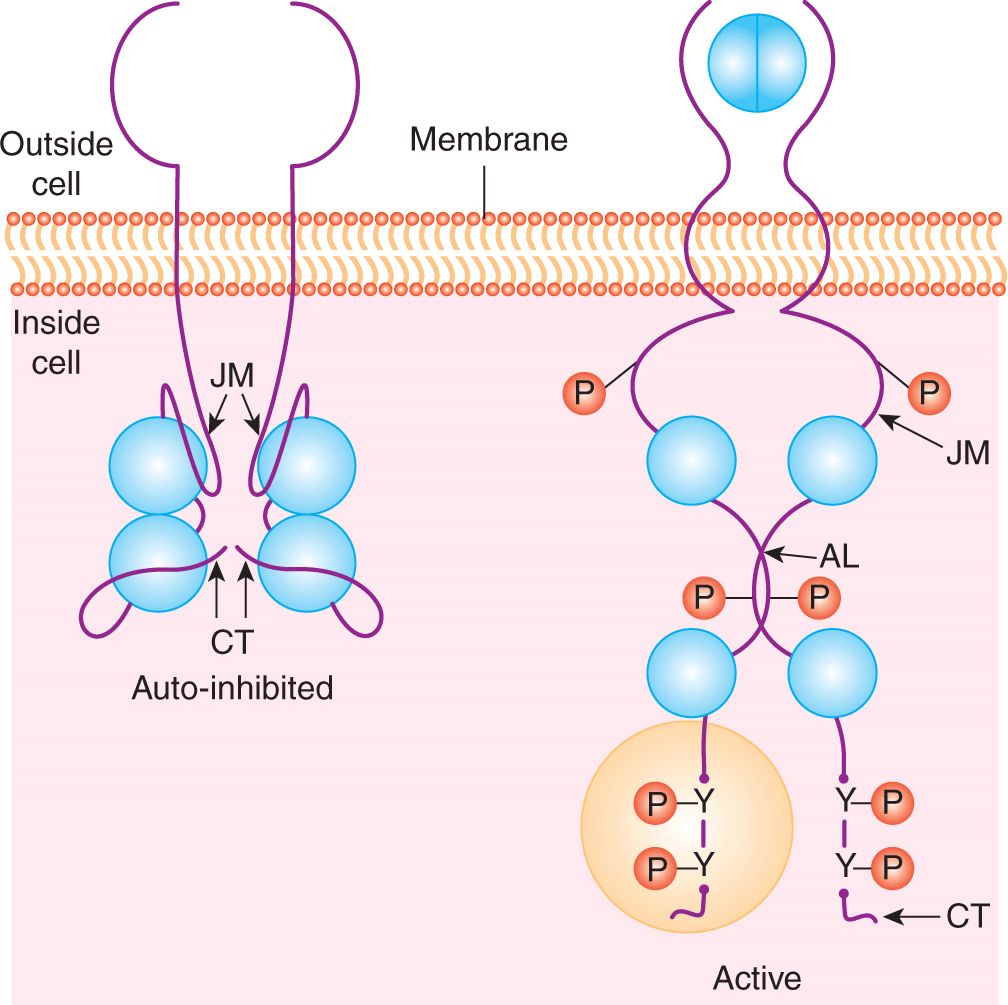
FIGURE 8–2 Growth factor receptor dimerization and activation. In the absence of ligand (eg, EGF) binding the intracellular kinase domain is inactive, held in a repressed conformation by intramolecular interactions involving the juxtamembrane region (JM), carboxyterminal tail (CT) and activation loop (AL). Ligand binding induces receptor dimerization, relief of inhibitory constraints and autophosphorylation of the intracellular domains on tyrosine residues. These autophosphorylation sites function to both enhance the catalytic activity and serve as docking sites for intracellular signaling molecules that bind to phosphotyrosine. P, Phosphate; Y, tyrosine.
Ligand binding and receptor dimerization bring together 2 catalytic domains, resulting in intermolecular autophosphorylation (transphosphorylation) of tyrosine residues within the catalytic domain and in the noncatalytic regulatory regions of the cytoplasmic domain. Phosphorylation of key residues within the kinase activation loop induces the opening of the catalytic site and allows access to ATP and protein substrates, while phosphorylated residues in noncatalytic regions create docking sites for downstream signaling molecules that are essential for signal propagation (see Fig. 8–2) (Lemmon and Schlessinger, 2010; Pawson, 2002).
Dimerization of receptors also leads to conformational changes within the cytoplasmic domain required for full catalytic activity. RPTKs are autoinhibited through intramolecular interactions that occlude the enzyme active site and prevent access of protein substrates. The juxtamembrane region of receptors from the PDGFR family represses the activity of the kinase domain and this repression is relieved by phosphorylation of tyrosine residues in the juxtamembrane region (Hubbard, 2004; Lemmon and Schlessinger, 2010). Similarly, the carboxyterminal tail of the angiopoietin receptor Tie2 is thought to block the active site of the kinase domain preventing substrate access (Niu et al, 2002).
Abnormal RPTKs involved in cancer are deregulated by loss of 1 or more of the regulatory mechanisms described above, making their catalytic activity ligand-independent. Identification of aberrantly activated RPTKs has led to the development of selectively targeted cancer therapeutics. For example, the ERBB2/NEU oncogene encodes a member of the EGFR family that is frequently amplified in human breast tumors (Slamon et al, 1987). Increased expression in this case is thought to increase the concentration of active dimers generating continuous and inappropriate cellular signaling. Trastuzumab, a humanized monoclonal antibody that targets the extracellular domain of ERBB2 (HER-2), is used in combination with chemotherapy to treat breast cancer and has shown improvement in survival of patients with ERBB2-positive tumors, when used as adjuvant therapy or for treatment of advanced disease (Hudis, 2007). Also, the small molecule tyrosine kinase inhibitors erlotinib and gefitinib significantly improve progression-free survival of patients with non–small cell lung cancer that harbor kinase domain-activating mutations of the EGF receptor (Cataldo et al, 2011; Lynch et al, 2004). Mutations in the juxtamembrane regulatory regions of both c-KIT and FLT3 have been implicated in gastrointestinal stromal tumors and acute myeloid leukemia, respectively, and have also been targeted with selective tyro-sine kinase inhibitors (Antonescu, 2011; Kindler et al, 2010). Although RPTKs remain attractive drug targets, the development of resistance through the acquisition of second site mutations of the oncogenic kinase, argues for therapeutic strategies that combine RPTK inhibition with targeting of downstream signaling pathways (Engelman and Settleman, 2008).
8.2.2 Formation of Multiprotein Complexes and Signal Transmission
Signaling pathways downstream of activated RPTKs are activated via interactions of specific proteins that create networks of signaling molecules. These signaling networks consist of both preformed and rapidly associating protein complexes that transmit information throughout the cell. A unifying feature of cytoplasmic signaling proteins is the presence of one or more conserved noncatalytic domains that mediate sequence-specific protein–protein interactions. The modular nature of these domains allows them to be used in diverse groups of cytoplasmic signaling molecules (Pawson and Nash, 2003). Many of these domains bind specifically to short (typically less than 10 amino acids) contiguous regions of their target protein. The binding of specific domains to their target sometimes requires phosphorylation of amino acids within the sequence-specific binding motif. Proteins that contain either SH2 (Src homology 2) or PTB (phosphotyrosine binding) domains, which recognize tyrosine phosphorylated sequence motifs are central to the formation of signaling complexes following activation of growth factor receptor tyrosine kinases (see Figs. 8–2 and 8–3) (Schlessinger and Lemmon, 2003).
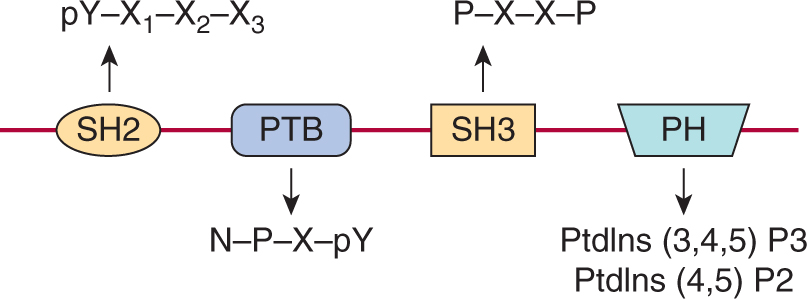
FIGURE 8–3 Examples of modular interaction domains. Representation of the protein modules commonly found in intracellular signaling proteins that are linked to growth factor receptor signaling cascades. Each is shown with its consensus peptide or phospholipid binding target. N, Asparagine; P, proline; X, any amino acid; Y, tyrosine. The domains are represented as if linked together on a single polypeptide to illustrate how the presence of multiple domains within signaling molecules would facilitate the assembly of larger signaling complexes. SH2, Src homology 2; SH3, Src homology 3; PTB, Phosphotyrosine binding; PH, Pleckstrin homology.
The SH2 domain was identified as a conserved region containing approximately 100 amino acids found outside the catalytic domain of Src family cytoplasmic tyrosine kinases. The specificity of SH2 domain recognition is determined both by the requirement for phosphotyrosine, common to almost all SH2 domains and by 3 to 4 amino acids (often termed the +1, +2, and +3 residues relative to the phosphotyrosine) on the carboxyterminal side of the tyrosine residue (see Fig. 8–3). Individual SH2 domains bind selectively to distinct phosphopeptide motifs, and the preferred consensus binding sequences for most SH2 domains have been defined. PTB domains can also specifically bind phosphotyrosine-containing peptides but, in contrast to SH2 domains, PTB domains recognize phosphotyrosine within a sequence motif that includes amino acids on the aminoterminal side of the tyrosine residue (see Fig. 8–3; Blaikie et al, 1994; Forman-Kay and Pawson, 1999).
Activation of growth factor receptors results in the auto-phosphorylation of the receptor at multiple tyrosine residues resulting in the creation of docking sites for cytoplasmic proteins that contain SH2 or PTB domains. Docking sites can also be created by the phosphorylation of cytoplasmic molecules, such as insulin receptor substrate-1 (IRS-1) by the insulin receptor. As such, phosphorylated IRS-1 becomes a docking site for SH2 domain-containing proteins (Myers et al, 1994). In this way SH2 and PTB domains play a crucial role in linking external signals received by a membrane receptor to cytoplasmic signaling pathways.
Since the original description of the SH2 domain, many additional protein modules have been identified, and their 3-dimensional structures have been described and distinct target specificities defined (Seet et al, 2006). All of these interaction modules represent independently folding domains with amino and carboxy termini in close proximity and a discrete surface ligand-binding interface, even when incorporated into a larger polypeptide. In addition to the tyrosinephosphorylated peptides described above, the specific binding partners for protein modules include phosphoserine- or phosphothreonine-containing peptides, proline-rich peptides, and carboxyterminal motifs, and membrane phospholipids.
Two additional protein interaction domains commonly found in signaling molecules downstream of RPTKs are the SH3 (Src homology 3) and PH (Pleckstrin homology) domain (see Fig. 8–3). SH3 domains are approximately 60 amino acids in length and are commonly found in signaling proteins in combination with other interaction molecules. These modules often bind to proline-based motifs in target proteins and the interaction is not dependent on changes induced by phosphorylation (Yu et al, 1992). SH3 domains are known to function both in the assembly of multiprotein complexes, and also as regulatory domains in intramolecular interactions. PH domains are protein modules of approximately 120 amino acids in length that interact specifically with membrane phosphoinositides (phosphorylated forms of phosphatidylinositol; PtdIns) (Harlan et al, 1994). Phosphoinositides are found at low levels within the cell and can be rapidly modified by phosphorylation in response to signaling. Importantly, PH domains recognize specific phosphoinositides such as PtdIns(3,4,5)P3 that are transiently produced following activation of growth factor receptors. Thus, an important function of PH domains is the recruitment of proteins to the membrane in the vicinity of an activated growth factor receptor (Seet et al, 2006; Lemmon and Schlessinger, 2010).
There is an additional class of signaling proteins that have no catalytic function (eg, NCK, CRK, and GRB2 [growth factor receptor bound-2]) and are composed entirely of SH2 and SH3 domains. These molecules are adaptor proteins that function by interacting with signaling enzymes that do not contain SH2 domains (or other phosphotyrosine containing modules such as PTB domains) thereby coupling them to a tyrosine kinase signaling complex. Each of these adaptor molecules has a different capacity to form protein complexes as a result of the binding specificity of its SH2 and SH3 domains, and the result is an organized but complex network of protein-protein interactions essential to coordinate an appropriate cellular response (Seet et al, 2006).
Figure 8–4 illustrates two examples of how protein modules function to activate growth factor receptor signal transduction. The SH2 and SH3 domain-containing adaptor protein, GRB2 plays a critical role in the activation of the small guanosine triphosphatase (GTPase) protein, RAS, a central transducer of growth factor receptor signals. As described below, RAS proteins are membrane-associated molecules that actively signal when bound to the guanine triphosphate nucleotide GTP. The SH2 domain of GRB2 associates with activated growth factor receptors while its SH3 domains are bound to proline-based motifs in SOS (son-of-sevenless), a guanine nucleotide exchange protein that activates RAS. Consequently, receptor activation leads to the recruitment of the GRB2-SOS complex close to its target, RAS, leading to its activation and downstream signaling (Lemmon and Schlessinger, 2010).
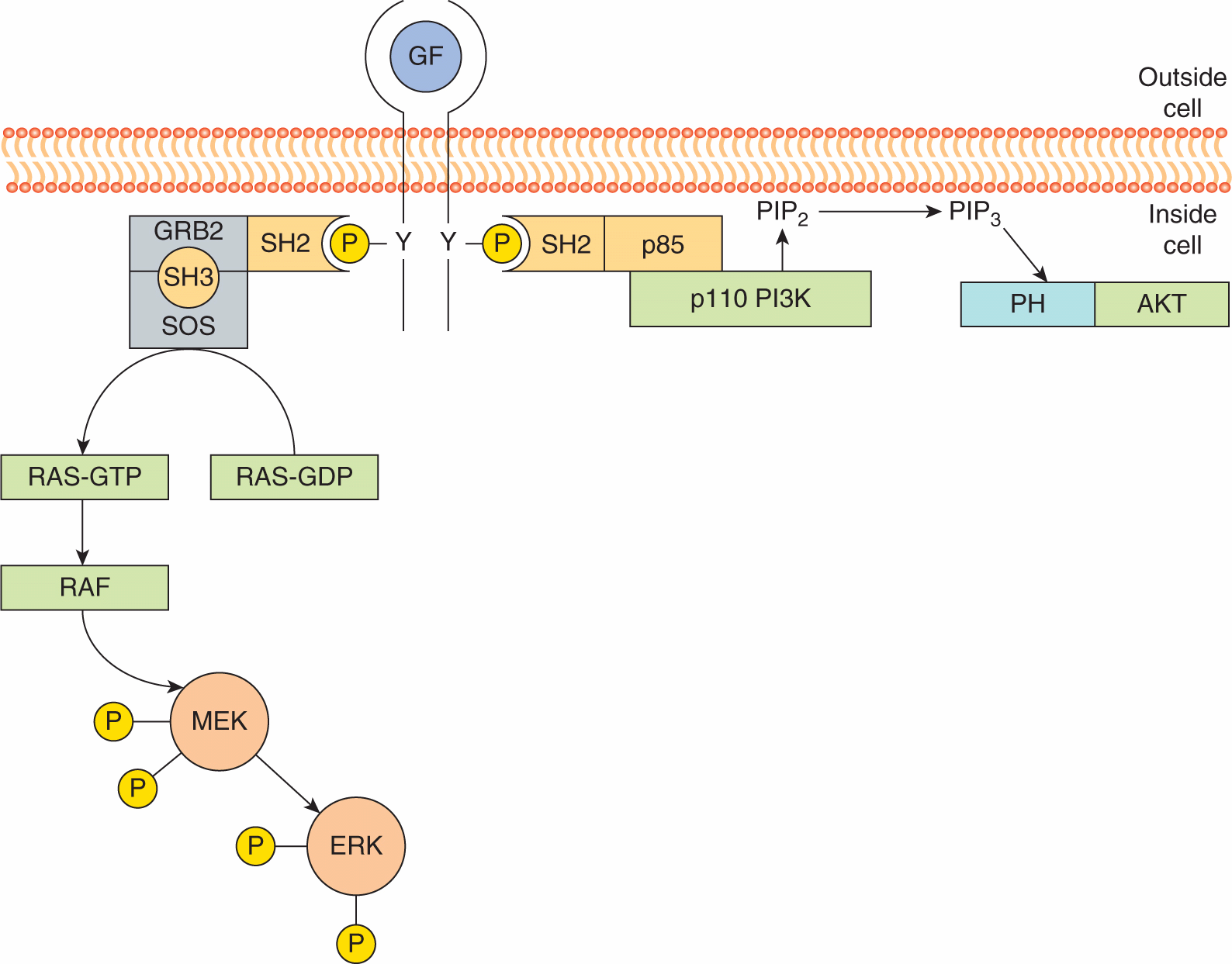
FIGURE 8–4 Recruitment of cytoplasmic signaling molecules by receptor protein tyrosine kinases. Binding of a receptor to a growth factor (GF) leads to phosphorylation of the intracellular domain on tyrosine (Y) residues; this interaction allows the SH2 domain-mediated association of enzymes and adaptor molecules such as GRB2 and p85. GRB2 is associated with the guanine nucleotide exchange factor SOS (son-of-sevenless). SOS recruited to activated receptor complexes then catalyzes the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on RAS. GTP-bound RAS binds the protein kinase RAF and activates a kinase cascade including MEK and ERK (see Sec. 8.2.4). The heterodimeric phosphoinositide kinase, phosphatidylinositol-3 kinase (PI3K), is comprised of a catalytic subunit, p110, and an adaptor or regulatory subunit, called p85 that contains two SH2 domains. Binding of the p85/p110 complex to activated growth factor receptors via the p85 SH2 domain activates catalytic activity of p110 which phosphorylates phosphotidylinositol 4,5 bisphosphate (PIP2). The product of this reaction, phosphatidylinositol 3,4,5 triphosphate (PIP3), serve as membrane anchoring site for PH domain-containing proteins such as AKT. See text for additional details.
Activation of growth factor receptors also results in the activation of phosphoinositide kinases that phosphorylate the 3′ hydroxyl group of the inositol ring. Phosphatidylinositol-3 kinase (PI3K) is a heterodimer made up of a catalytic subunit, p110, and a regulatory subunit, p85 that contains 2 SH2 domains. Following receptor activation, the PI3K is recruited to the activated receptor by the p85 SH2 domains, which binds to specific phosphotyrosine interaction motifs, leading to allosteric activation of the p110 catalytic subunit and the production of PtdIns(3,4,5)P3 (described in Sec. 8.2.5; Lemmon and Schlessinger, 2010).
8.2.3 RAS Proteins
RAS proteins control signaling pathways that regulate normal cell growth and malignant transformation. Three human RAS genes encode the proteins H-RAS, K-RAS and N-RAS and are part of a large family of low-molecular-weight GTP-binding proteins. RAS proteins have a molecular weight of 21 kDa (hence the designation p21ras) and share 85% sequence homology. The RAS proteins are GTPases that cycle between an active GTP-bound “on” and an inactive guanosine diphosphate (GDP)-bound “off” configuration in response to extracellular signals, essentially functioning as a molecular binary switch (Fig. 8–5). RAS is activated by the effects of guanine nucleotide exchange factors (GEFs), such as SOS described above, that releases RAS-bound GDP and allows GTP binding to RAS (Buday and Downward, 2008).
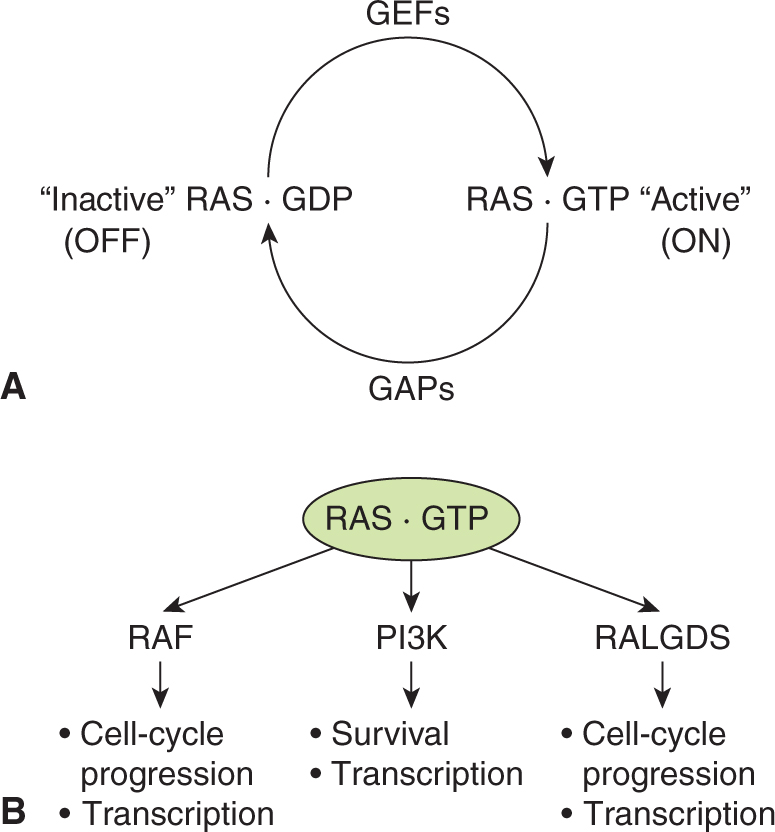
FIGURE 8–5 RAS protein activation and downstream signaling. A) The small GTPase RAS cycles between an inactive GDP bound state and the active GTP bound state. RAS activation is regulated by guanine nucleotide exchange factors (GEFs) that promote exchange of GDP for GTP. GTP hydrolysis requires GTPase-activating proteins (GAPs) that enhance the weak intrinsic GTPase activity of RAS proteins. B) Once in its active GTP bound form RAS interacts with different families of effector proteins including RAF protein kinases, PI3Ks and RAL GDS, a GEF for the RAS-related protein RAL. Activation of these downstream pathways leads to cellular responses including gene transcription, cell-cycle progression and survival.
In the active GTP-bound form, RAS binds to a number of distinct effector proteins that, in turn, activate downstream signaling cascades. One of the best characterized effectors is the protein kinase RAF. RAS-GTP binding to RAF activates its kinase activity, and consequently activates a downstream cascade of protein kinases that include MEK and ERK (see Fig. 8–4). Additional RAS-GTP effectors include the exchange factor for another small GTPase RAL (RALGDS), and the p110 catalytic subunit of PI3K (see Fig. 8–5). Through these diverse effectors RAS proteins regulate cell-cycle progression, cell survival, and cytoskeletal organization (Reuther and Der, 2000).
Termination of RAS activity occurs through the hydrolysis of GTP, converting it to GDP by action of GTPase-activating proteins (GAPs) that promote the intrinsic GTPase activity of the RAS proteins themselves. Therefore, a balance to the activities of GEFs and GAPs determines the activity of normal RAS proteins. Both the GEFs and a number of the GAP family members, which are often represented by p120GAP, are themselves regulated by receptor tyrosine kinase signaling cascades (Wittinghofer, 1998).
The normal function of RAS proteins requires posttranslational modification. Newly synthesized RAS proteins are modified by the addition of a lipid chain to a cysteine residue in the carboxy terminus of RAS proteins. This covalently linked lipid is either a farnesyl or geranylgeranyl group (collectively termed prenylation) and is required for RAS association with intracellular membranes. It is important for the oncogenic activity of RAS proteins. Both H-RAS and N-RAS are also subsequently modified by the addition of 2 palmitoyl long-chain fatty acids important for the correct localization of these proteins to specific parts of the membrane. RAS proteins are activated and signal from specific microdomains within the plasma membrane, as well as distinct subcellular compartments such as the Golgi and endosomes (Hancock, 2003).
Abnormalities in RAS protein activity have been identified in greater than 30% of human malignancies as a result of either mutations (most commonly in K-RAS) that render it locked in an active GTP-bound state, or activated as a result of deregulated signaling from upstream pathways (see Chap. 7, Sec. 7.5.5). RAS proteins have been targets for anticancer therapeutics. Because prenylation involves the activity of a protein farnesyl transferase or a geranylgeranyl transferase, inhibitors of these enzymes were developed as potential inhibitors of oncogenic RAS activity (Berndt et al, 2011). Farnesyl transferase inhibitors (FTIs) were demonstrated to be potent killers of tumor cells in culture and in animal models, but results of clinical trials showed no survival advantages for most patients with solid or hematological malignancies. Inhibitors of geranylgeranyl transferase inhibitor (GTI) have shown similarly promising in vitro properties and are being evaluated in clinical trials. The efficacy of FTIs does not depend on the presence of activating RAS mutations and may be linked to the prediction that hundreds of proteins are modified by prenylation. While FTIs and GTIs were developed to inhibit RAS activity, other prenylated proteins, such as Rheb, an activator of TORC1 (described in Sec. 8.2.5), may be important targets of FTI activity in tumors. The effective use of these agents in cancer therapy will require the identification of biomarkers, perhaps the farnesyl transferase substrates themselves, of response to FTIs (Berndt et al, 2011).
8.2.4 Mitogen-Activated Protein Kinase Signaling Pathways
Mitogen-activated protein kinases (MAPKs) control highly conserved signaling pathways that regulate all eukaryotic cells. Mammalian cells contain multiple distinct MAPK pathways that respond to divergent signals including growth factors and environmental stresses such as osmotic stress and ionizing radiation. All MAPK pathways include a core 3-tiered signaling unit, in which MAPKs are activated by the sequential activation of linked serine/threonine kinases (Fig. 8–6). The MAPK pathway is activated by phosphorylation of threonine and tyrosine residues in a T-X-Y (T = threonine, X = any amino acid, Y = tyrosine) motif in the kinase activation loop. This phosphorylation is achieved by a family of dual specificity kinases referred to as MEKs or MKKs (MAPK-kinase). MEK (mitogen-activated protein [MAP]/extracellular signal-related kinase [ERK] kinase) activity is regulated by serine and threonine phosphorylation catalyzed by kinases called MAP3Ks (MAPK-kinase-kinase). A number of distinct families of MAP3Ks are activated by diverse upstream stimuli that link the activation of the MAPK signaling unit to extracellular signals. These structurally related pathways are controlled by stimuli that elicit very distinct physiological consequences (ie, mitogenesis or the stress response). Within each pathway specificity is determined by scaffold molecules that link specific core components. Similarly, although all MAPKs phosphorylate very similar consensus motifs in their target substrates, specificity of protein substrate selection is ensured by docking domains that mediate binding of specific kinases to their substrates (Sharrocks et al, 2000).
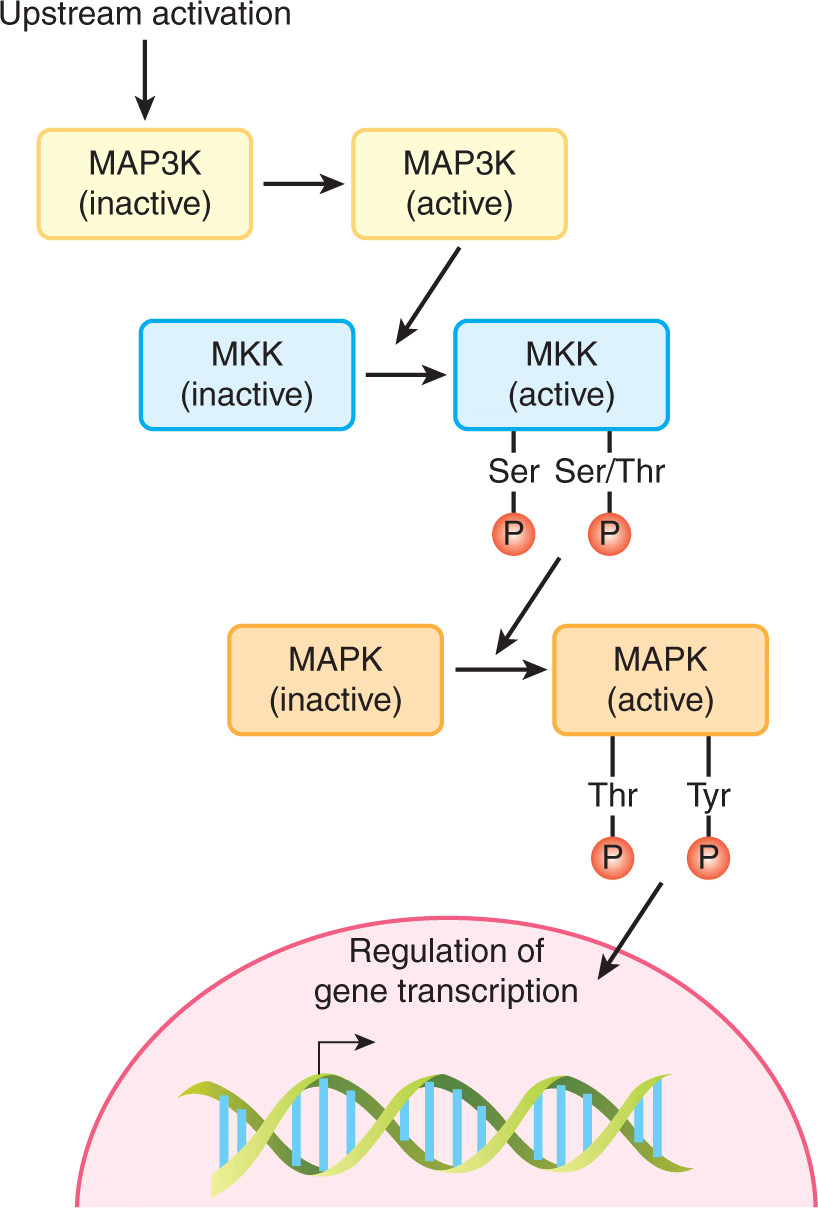
FIGURE 8–6 The MAPK core signaling module. All MAPK pathways include a core 3-tiered signaling unit, in which MAPKs are activated by the sequential activation of linked serine/threonine kinases. The MKKs are unique dual specificity kinases that phosphorylate both tyrosine (Y) and threonine (T) residues within an activation motif found in the MAPKs. P, Phosphate.
Three distinct MAPK pathways have been characterized in mammalian cells: the extracellular signal regulated kinase 1 and 2 (ERK1/2), the c-Jun N-terminal kinase or stress-activated protein kinase (JNK/SAPK), and p38. As described in Section 8.2.3, activation of RAS proteins causes the activation of RAF, a MAP3K upstream of ERK1/2. ERK kinase activation is a final signaling step that is shared amongst several pathways stimulated by growth factor receptors, such as those for EGF, PDGF, FGF (see Chap. 7, Sec. 7.5.3), and by more diverse stimuli from cytokine receptors, and antigen receptors (Katz et al, 2007). RAF directly activates MEK-1/2 by phosphorylating it on serine residues, which enhances the availability of the catalytic site to potential substrates. Activated MEK-1/2 is a dual-specificity kinase that phosphorylates the ERK kinases. MEK-induced phosphorylation of ERK occurs on threonine and tyrosine residues in the activation loop, which induces catalytic activation of ERK and phosphorylation of both cytoplasmic and nuclear protein substrates that regulate cell migration, proliferation, and differentiation. In the cytoplasm, activated ERK phosphorylates cytoskeletal proteins as well as the RSK family of protein kinases. Activated RSK kinases regulate translation, transcription, and survival signaling through phosphorylation of both nuclear and cytoplasmic substrates (Anjum and Blenis, 2008). Activation of ERK also induces its translocation to the nucleus where it phosphorylates and activates transcription factors (see Sec. 8.2.6), including SP1, ELK-1 and AP-1 (comprised of FOS and JUN) (Fig. 8–6).
Given the broad biological outcomes of MAPK signaling, it is not surprising that dysregulation of these pathways has been implicated in malignant transformation. Increased levels of activated ERKs are frequently found in human tumors, and often are attributable to the presence of mutations in RAS or other upstream components in growth factor signaling cascades. Activating mutations in the RAF family member BRAF, occur in 70% of malignant melanoma and at lower frequency in a wide range of other human tumors (see also Chap. 7, Sec. 7.5.5; Davies et al, 2002). The most common mutation, V600E, is a valine-to-glutamic-acid substitution in the activation loop of the kinase domain that results in constitutive activation. In Phase III clinical trials in metastatic melanoma, the selective BRAF inhibitor vemurafenib has produced tumor regression and improved survival in patients with V600E BRAF mutations (Chapman et al, 2011).
8.2.5 Phosphoinositide Signaling
Phosphoinositides are rare phospholipids of cell membranes that are dynamically regulated in response to growth factor signaling. They contribute to signal propagation by 2 main mechanisms; by serving as precursors of the second messengers diacylglycerol (DAG) and inositol triphosphate (Ins(1,4,5)P3) and Ca2+, or by binding to signaling proteins that contain specific phosphoinositide binding modules. Figure 8–7 illustrates some of the important phospholipid products that function in growth factor signal transduction pathways. Phosphoinositides can be phosphorylated or dephosphorylated by lipid kinases and phosphatases at distinct positions on the inositol ring in response to growth factor signaling. Activation of PI3K (described in Sec. 8.2.2), which specifically phosphorylates the 3′ position, leads to the rapid production of PtdIns(3,4,5)P3. Levels of PtdIns(3,4,5) P3 are tightly controlled by the action of inositol phosphatases. Phosphatase and tensin homolog (PTEN) is a 3′-phosphoinositide phosphatase that dephosphorylates the 3′ position of PtdIns(3,4,5)P3 and PtdIns(3,4)P2, and therefore functions as a major negative regulator of PI3K signaling (see Chap. 7, Sec. 7.6.2).
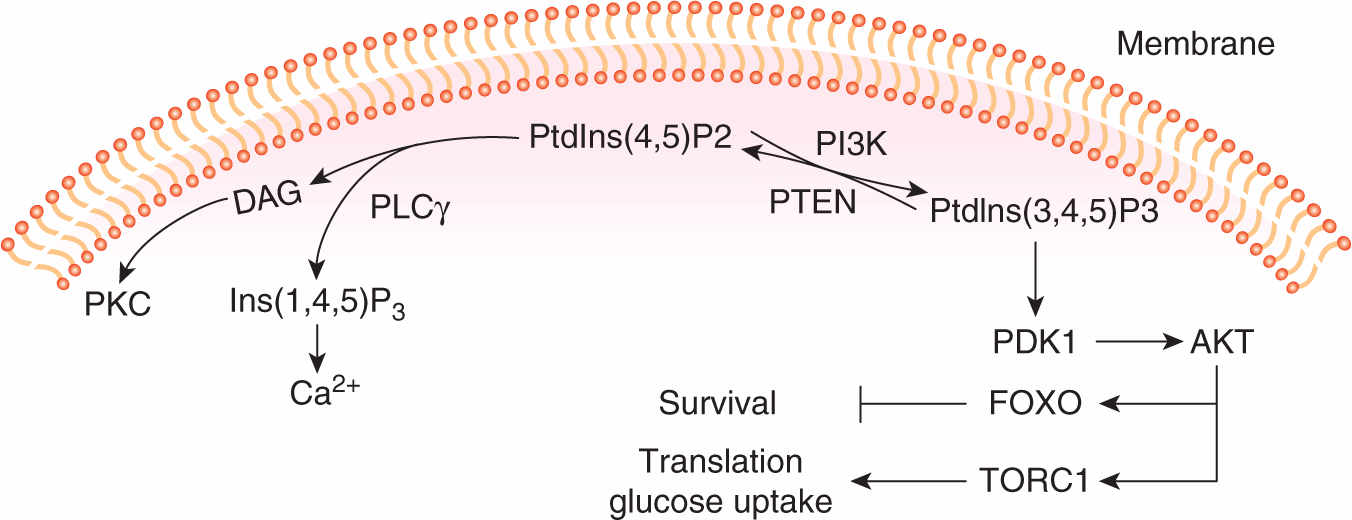
FIGURE 8–7 Phosphoinositide metabolism in growth factor signaling. The positions in the inositol ring that are modified by phosphorylation are numbered. PtdIns(4,5)P2 is hydrolyzed by phospholipase Cγ (PLCγ) to produce second messengers DAG (diacylglycerol) and inositol triphosphate (Ins(1,4,5)P3), that results in activation of Protein Kinase C (PKC) and release of calcium (Ca2+) from intracellular stores. Lipid kinases (PI3K) and phosphatases (PTEN) positively or negatively regulate the phosphorylation of the inositol 3′ position and the production of PtdIns(3,4,5) P3. PtdIns(3,4,5) signals downstream to activate PDK1 and AKT and the activation of metabolic and survival pathways (see text for details).
The production of PtdIns(3,4,5)P3 leads to the recruitment of the PH-domain-containing protein serine/threonine kinases PDK1 and AKT. AKT is activated by conformational changes evoked by phospholipid binding, and phosphorylation by PDK1 at threonine 308. Full activation of AKT requires phosphorylation at a second site (serine 473) by the serine kinase complex TORC2. A number of important substrates for activated AKT have been identified that fall into 2 main classes as regulators of cell survival or regulators of cell proliferation (see Chap. 9, Sec. 9.2.4). Briefly, AKT phosphorylates substrates that, in turn, lead to the activation of TORC1, the serine/threonine kinase mammalian target of rapamycin (mTOR) complex (see below) that regulates cell functions required for growth such as protein translation, glucose uptake, and glycolysis (Zoncu et al, 2011). Activated AKT also controls cell survival through the phosphorylation and nuclear exclusion of the FOXO transcription factors, preventing the expression of genes that can induce cell death (Manning and Cantley, 2007).
Mammalian target of rapamycin (mTOR) is a serine threonine kinase that interacts with additional proteins to form two distinct functional complexes, termed mTOR Complex 1 (TORC1) and 2 (TORC2). TORC1 controls a wide range of cellular processes that promote cell proliferation (see Chap. 9, Sec. 9.2.4) including protein synthesis through the phosphorylation of eukaryotic translation initiation factor binding protein (4E-BP1) and S6 kinase 1 (S6K1) that in turn promote cap-dependent protein translation. TORC1 regulates ATP production and metabolism by promoting the expression of hypoxia-inducible factor 1a (HIF1a) that regulates the expression of glycolytic genes controlling glucose metabolism (see Chap. 12, Sec. 12.2.3). TORC1 also inhibits autophagy (see Chap. 12, Sec. 12.3.6), a process required for cellular catabolism under nutrient starvation, by phosphorylation and inhibition of the activity of autophagy-related gene 13 (ATG13), which forms part of a kinase complex required to initiate autophagy. Much less is known about the functions of TORC2 but its activity also promotes cell growth through activation of protein kinases AKT (described above), serum- and glucocorticoid-induced kinase (SGK1), and protein kinase C-a (PKCa). The SGK1 kinase is activated by TORC2 and regulates ion transport and cell growth. TORC2 can also regulate actin cytoskeleton dynamics through the phosphorylation and activation of PKCa (Laplante and Sabatini, 2012).
Human malignancies are frequently associated with inactivating mutations in the PTEN gene (see Chap. 7, Sec. 7.6.2; Li et al, 1997). Loss of PTEN leads to accumulation of 3′-phosphinositides, causing deregulated AKT activity and malignant transformation (Stambolic et al, 1998). High-throughput sequencing has identified activating mutations in PI3K-CA, the p110 subunit of PI3K in a wide variety of tumors, including 25% to 40% of breast tumors, making it the most commonly mutated gene in breast cancer (Samuels and Waldman, 2010). PI3K-CA inhibitors and AKT inhibitors are being developed and tested in clinical trials, but associated toxicity has been substantial. Inhibition of mTOR (TORC1) has been more successful as a therapeutic strategy. The mTOR inhibitor, rapamycin, was originally developed as an anti-fungal agent and subsequently has been used as an immunosuppressant. Rapamycin analogs temsirolimus and everolimus have been used to treat renal cell carcinoma and other human tumors.
8.2.6 Transcriptional Response to Signaling
One important consequence of growth factor signaling is the transcription of genes that coordinate cell growth, cellular differentiation, cell death, and other biological effects. Transcription of genes is catalyzed by the enzyme RNA polymerase II and regulated by supporting molecules, collectively termed transcription factors. Transcription factors can activate or repress gene expression by binding to specific DNA recognition sequences, typically 6 to 8 base pairs in length, found in the promoter regions at the 5′ end of genes. The formation of RNA transcripts is influenced by the interaction of these gene-specific factors with elements of a common core of molecules regulating the activity of RNA polymerase II (Woychik and Hampsey, 2002). The activity of transcription factors can be modified, frequently by phosphorylation, through the activity of many of the signaling pathways described above, including MAPK and the PI3K pathways, which can act in the nucleus to directly modify transcription factor activity (Brivanlou and Darnell, 2002).
Transcription factors are modular, consisting of a specific DNA binding region that binds to specific DNA sequences, as described below, and an activation or repression domain, which interacts with other proteins to stimulate or repress transcription from a nearby promoter. Based on the structure of their DNA-binding domains, transcription factors can be placed into homeodomain (sometimes called helix-turn-helix), zinc-finger, or leucine-zipper, and helix-loop-helix (HLH), groupings (Fig. 8–8).
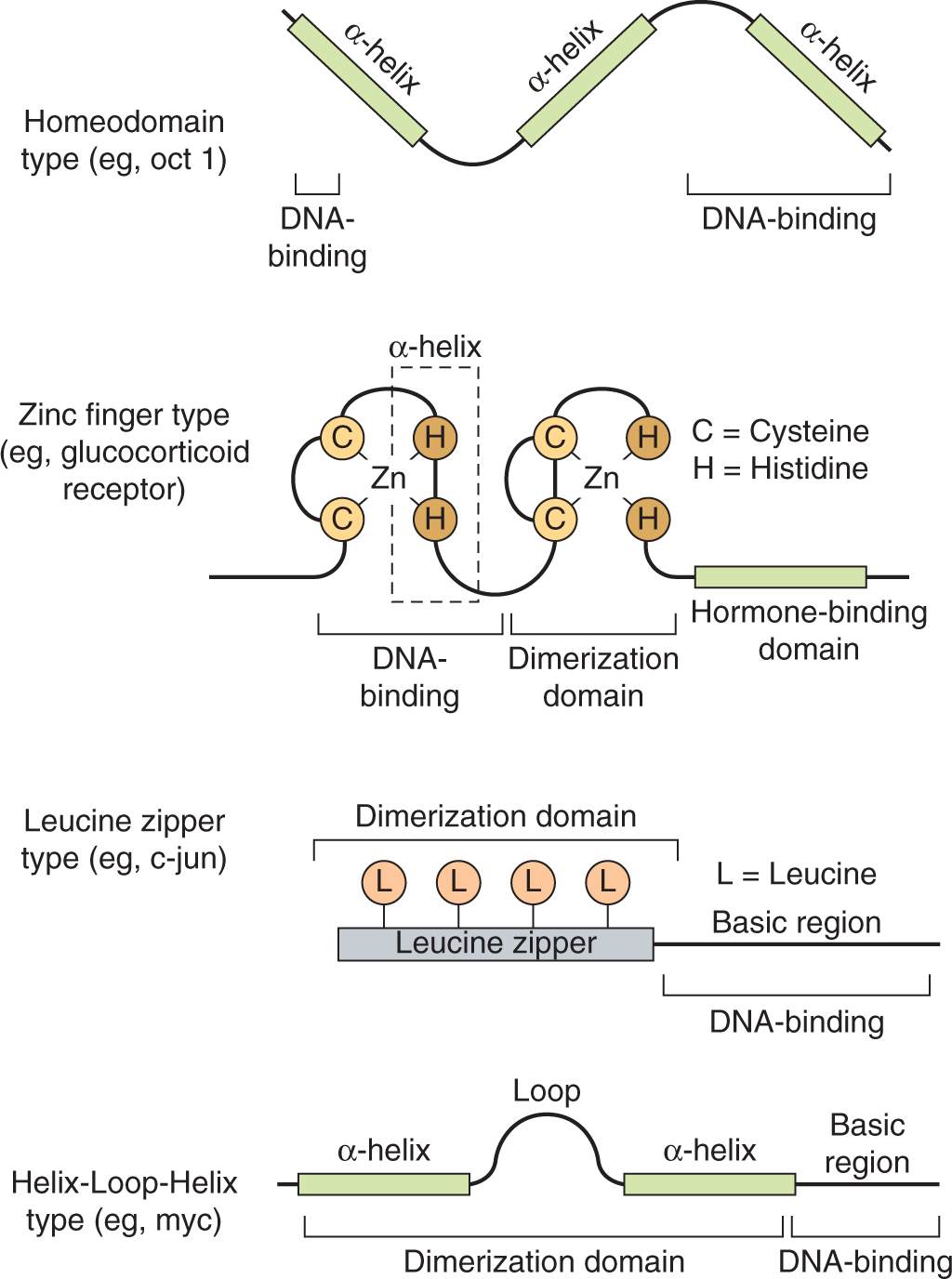
FIGURE 8–8 Transcription factor DNA-binding domain classes. The general structure of the 4 major classes of DNA-binding domains found in modular transcription factors is illustrated.
The homeodomain factors contain a 60-amino-acid DNA-binding domain called a homeobox that is similar to the helix-turn-helix domain first described in bacterial repressors. The name is derived from the Drosophila homeotic genes that determine body structure identity. In vertebrates, homeodomain proteins have similar properties and function as master regulators during development. The homeodomain contains 3 helical regions. The third helical region, as well as amino acids at the aminoterminal end of the homeodomain, directly contact DNA.
Zinc-finger transcription factors contain a sequence of 20 to 30 amino acids with 2 paired cysteine or histidine residues that are coordinated by a zinc ion. Binding of the zinc ion folds these polypeptide sequences into compact domains with α helices that insert into the DNA (see Fig. 8–8). Members of this group of transcription factors mediate differentiation and growth signals, including those caused by binding of steroid hormones to receptors; they have been implicated in malignancy.
Leucine-zipper transcription factors contain helical regions with leucine residues occurring at every seventh amino acid, which all protrude from the same side of the α-helix. These leucines form a hydrophobic interaction surface with leucine zippers of similar proteins. Additional members of this family contain other hydrophobic amino acids in the α-helices that make up the dimerization domain. The DNA binding regions of the α-helices in either case contain basic amino acids that interact with the DNA backbone. These factors, also referred to as basic zipper proteins, bind to DNA as homo- or heterodimers, and include the fos/jun pair (called the AP1 transcription factor), which becomes activated by cellular stress. Members of this group also tend to become activated by proliferative and developmental stimuli.
Basic helix-loop-helix factors are similar to the basic zipper factors described above, but include a loop region that separates the 2 α-helical regions of the polypeptide. The carboxyterminal α-helix mediates formation of homo- or heterodimers that contact DNA with basic amino acids found in the aminoterminal helix.
Activation and repression domains are structurally diverse regions, ranging from the random coil conformation of acidic activation domains to the highly structured ligand-binding domains of hormone receptors. Both transcription activators and repressors exert their effects by binding to multisubunit coactivators or corepressors that act to modify chromatin structure and assembly of RNA Pol II complexes. Enzymes that regulate histone acetylation and phosphorylation are key components of transcriptional activator and repressor complexes. Histone acetylation near the promoter regions of genes facilitates the interaction of the DNA with transcription factors while deacetylation results in condensed chromatin structures that inhibit assembly of the transcription machinery at the promoter (Berger, 2007).
Alteration in transcription factor function, which can cause unregulated activation and expression of genes, or lead to inappropriate repression of others, can lead to transformation and is well documented in human cancers. Although we understand how mutation or overexpression of transcription factors such as MYC and TP53 alters their activity, we do not yet fully understand how these changes influence the gene expression or repression patterns that bring about the oncogenic state.
8.2.7 Biological Outcomes of Growth Factor Signaling
Growth factor signaling results in changes in gene expression of large numbers of genes that program the physiological responses such as cell-cycle progression, cellular differentiation, cell growth, survival, or apoptosis. Changes in gene expression downstream of growth factor signaling proceeds in 2 stages. Expression of immediate early genes, which often encode transcription factors, does not require new protein synthesis. Immediate early gene expression is followed by expression of other genes, sometimes called delayed response genes, which are often the products of the transcription induced by the immediate early genes (Hill and Treisman, 1999).
Cells in the G1 phase of the cell cycle respond to external stimuli by either withdrawing from the cell cycle (Go) or advancing through the restriction point (see Chap. 9, Sec. 9.2.1) toward cell division. Progression through G1 and entry into S phase normally requires stimulation by mitogens, such as growth factors. For example the D-type cyclins are expressed as part of the delayed early response to stimulation of growth factor signaling cascades. These D-type cyclins assemble with cyclin-dependent kinases, and the active complex phosphorylates and inactivates the retinoblastoma (Rb) protein, releasing the E2F transcription factor family that in turn activate the transcription of genes required for S-phase entry (see Chap. 9, Sec. 9.2.3). A common property of cancer cells is their ability to undergo G1 phase progression in the absence of external mitogenic stimuli. Activating mutations in any of the growth factor signaling components upstream of G1 checkpoint control can lead to cyclin D accumulation, which drives continuous cell cycling (Evan and Vousden, 2001; Sherr, 1996).
A second important consequence of growth factor signaling is cell survival. Normal cells require continuous exposure to survival factors, such as soluble growth factors, or cell matrix interactions, to suppress apoptosis. Tissue homeostasis is maintained through the limited supply or spatial restriction of these factors that limit cell expansion. Evasion of this control mechanism is another common feature of tumor cells. Activating mutations in survival pathways, such as activating mutations in the PI3K pathway or loss of function mutations in PTEN, can confer resistance to apoptotic signals that would normally limit deregulated cell proliferation (Evan and Vousden, 2001).
8.2.8 Suppression of Growth Factor Signaling
Signaling from activated growth factor receptors is tightly regulated both temporally and spatially. Many proteins that antagonize receptor tyrosine kinase signaling are also recruited to active receptor complexes through interactions with SH2, PTB, or TKB domains. For example, the opposing action of protein tyrosine phosphatases can eliminate docking sites for proteins containing SH2 domains or inhibit tyrosine kinase activity by dephosphorylation of regulatory phosphorylation sites in the kinase activation loop (Chernoff, 1999; Tiganis, 2002). Similarly the action of lipid phosphatases such as PTEN (described in Sec. 8.2.5) function to antagonize PI3K-AKT signaling.
Modification of proteins with ubiquitin is an important mechanism for signal termination. Ubiquitination describes the covalent attachment of 1 or more 76-amino-acid ubiquitin molecules to a target protein. Ubiquitin modification involves a multistep process in which free ubiquitin is first attached to an ubiquitin-activating enzyme (E1) and subsequently transferred to an ubiquitin-conjugating enzyme (E2), which, in partner with an ubiquitin ligase (E3), transfers ubiquitin to the specific protein substrate (Hershko and Ciechanover, 1998; Fig. 8–9). The specificity of this process is determined by the E3 ligase that selectively binds substrates (Deshaies and Joazeiro, 2009; Rotin and Kumar, 2009). The consequences of protein modification by ubiquitin can include degradation by the 26S proteasome, which is a mechanism for eliminating activated enzymes in the cytosolic compartment (see Chap. 9, Sec. 9.2.2). Ubiquitination of transmembrane proteins such as activated RPTKs is followed by their endocytosis and transfer of activated receptors to the lysosome where they are degraded. This mechanism allows signal termination and return to a basal state after receiving and responding to growth factor signals.
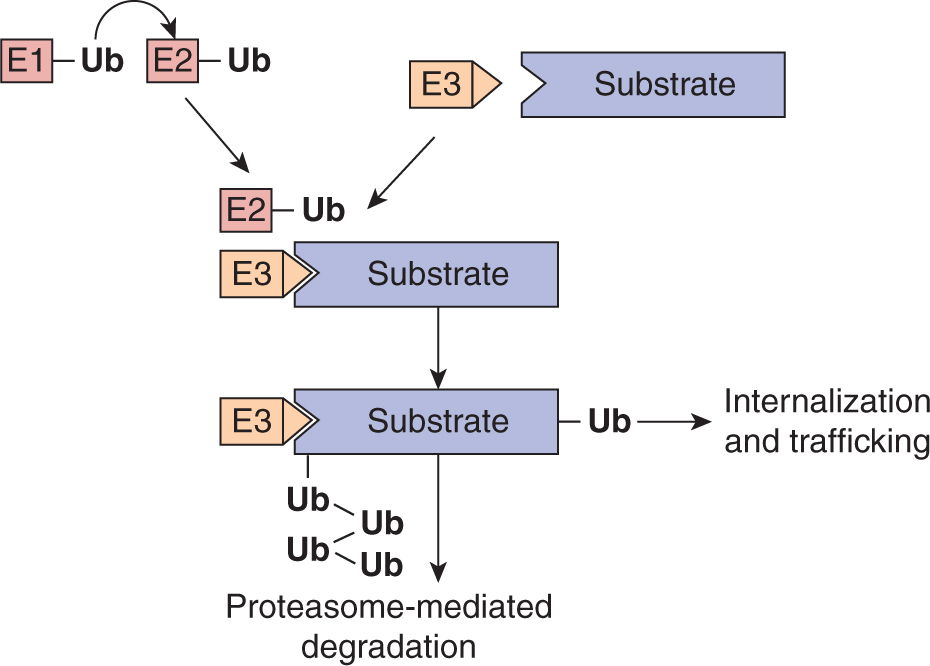
FIGURE 8–9 Ubiquitination pathway. Protein ubiquitination is a multistep process in which free ubiquitin is first attached to an ubiquitin-activating enzyme (E1) and subsequently transferred to an ubiquitin-conjugating enzyme (E2), which, in partner with an ubiquitin ligase (E3), transfers ubiquitin to the specific protein substrate. The E3 ligase provides specificity to the system by functioning as an adaptor to selectively bind substrates. Polyubiquitinated substrates are targeted for degradation by the proteasome while monoubiquitination of a substrate can serve as a signal for receptor internalization and trafficking to multivesicular bodies and the lysosome.
Failure of ubiquitination of RPTKs can result in prolonged activation and oncogenic signaling (Lu and Hunter, 2009). First identified as a transforming viral oncogene, c-CBL is an E3 ligase recruited to activated RPTKs through its phosphotyrosine binding TKB domain that promotes receptor ubiquitination. Mutant forms of c-CBL that lack ubiquitin ligase activity have been identified in myeloid malignancies and lung cancer (Kales et al, 2010; Tan et al, 2010). Although the mechanisms that drive oncogenic transformation by c-CBL are not fully understood, expression of c-CBL proteins that are devoid of E3 ligase activity promotes signaling downstream of activated tyrosine kinases (Kales et al, 2010).
8.3 CYTOPLASMIC TYROSINE KINASE SIGNALING
Tyrosine kinases (TKs) also play a central role in the transmission of signals from distinct classes of cell-surface receptors that themselves do not possess intrinsic TK activity. Cytoplasmic TKs are recruited to cell-surface molecules following receptor activation, and many of the intracellular events that occur resemble those evoked by receptor TKs as described in Section 8.2. Figure 8–10 is a schematic showing selected cytoplasmic TKs. A common feature of these cytoplasmic TKs is the presence of conserved protein modules, such as SH2 and SH3 domains. These protein modules function to regulate kinase activity and also serve to couple these molecules to extracellular receptors. Some of the features of receptor signaling pathways that utilize cytoplasmic TKs are described below.
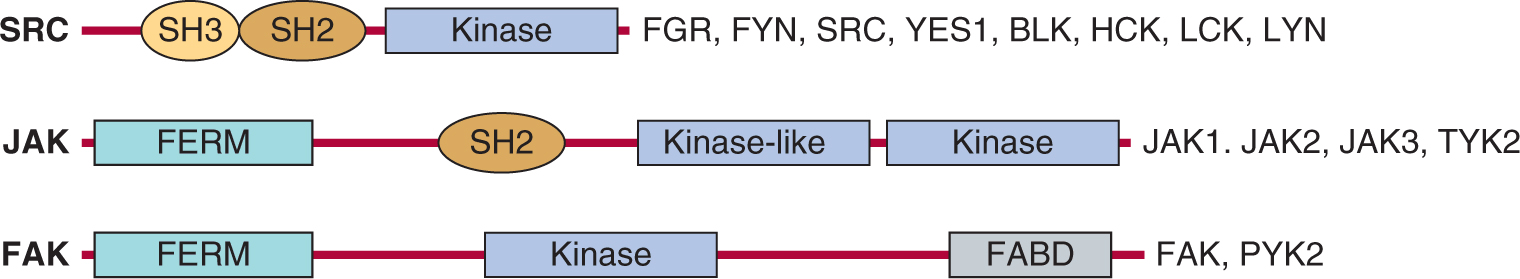
FIGURE 8–10 Cytoplasmic protein TKs. The general structure of several cytoplasmic protein TK families is illustrated. Members of each family are listed beside each diagram.
8.3.1 Cytokine Signaling
Cytokines regulate the proliferation, differentiation and activity of hematopoietic cell lineages through interaction with structurally and functionally related receptors of the cytokine receptor superfamily. These receptors do not contain intrinsic TK activity, but rather transmit intracellular signals through the association with the Janus kinase (JAK) family of kinases. With the exception of the receptors for granulocyte colony-stimulating factor (G-CSF) and erythropoietin (see Chap. 12, Sec. 12.1.3.1), most cytokine receptors are multisubunit complexes and contain a unique ligand binding subunit and a common or shared signaling subunit. There are 4 distinct signaling chains: the gp130 subunit (interleukin [IL]-6Rβc, where “c” denotes the chain, is common to other cytokine receptors), and the IL-3Rβc, IL-2Rβc, and IL-2Rγc chains that can transmit signals from many different ligands.
Similar to RPTK signaling, cytokine binding leads to initiation of downstream signaling events (Fig. 8–11). As the cytoplasmic domains of the common receptor subunits do not possess intrinsic TK activity, noncovalent association with the JAK family of cytoplasmic TKs is required for signal transmission (Ihle, 1995; O’Shea et al, 2002). The JAKs contain a F for 4.1 protein, E for ezrin, R for radixin and M for moesin (FERM) domain, an SH2 domain, a functional TK domain, and a kinase-like domain (or pseudokinase domain) that can exert autoinhibitory activity on the kinase domain (see Fig. 8–10). The FERM domain functions to both regulate JAK kinase activity and mediate association with receptors, while the function of the JAK SH2 domain is unclear as it does not appear to bind to tyrosine phosphorylated proteins. Upon cytokine binding, JAKs are tyrosine phosphorylated and activated, leading to phosphorylation of specific tyrosine residues on the receptor. This activated complex generates docking sites for SH2-domain containing proteins such as the STAT (signal transducers and activators of transcription) family of transcription factors (Fig. 8–11) as well as GRB2 and PI3K (described in Sec. 8.2.3). STATs are then tyrosine phosphorylated, inducing homo- or heterodimerization and subsequent transport to the nucleus where they regulate gene expression. STATs contain a conserved aminoterminal DNA binding domain that is specific to the STAT family of proteins, a conserved SH2 domain, and a carboxyterminal tyrosine residue that mediates dimerization. In addition to tyrosine phosphorylation, STATs can be phosphorylated by serine/threonine kinases such as ERK and mTOR (see Sec. 8.2.5), and serine phosphorylation of STAT regulates its ability to activate transcription (Jatiani et al, 2010).
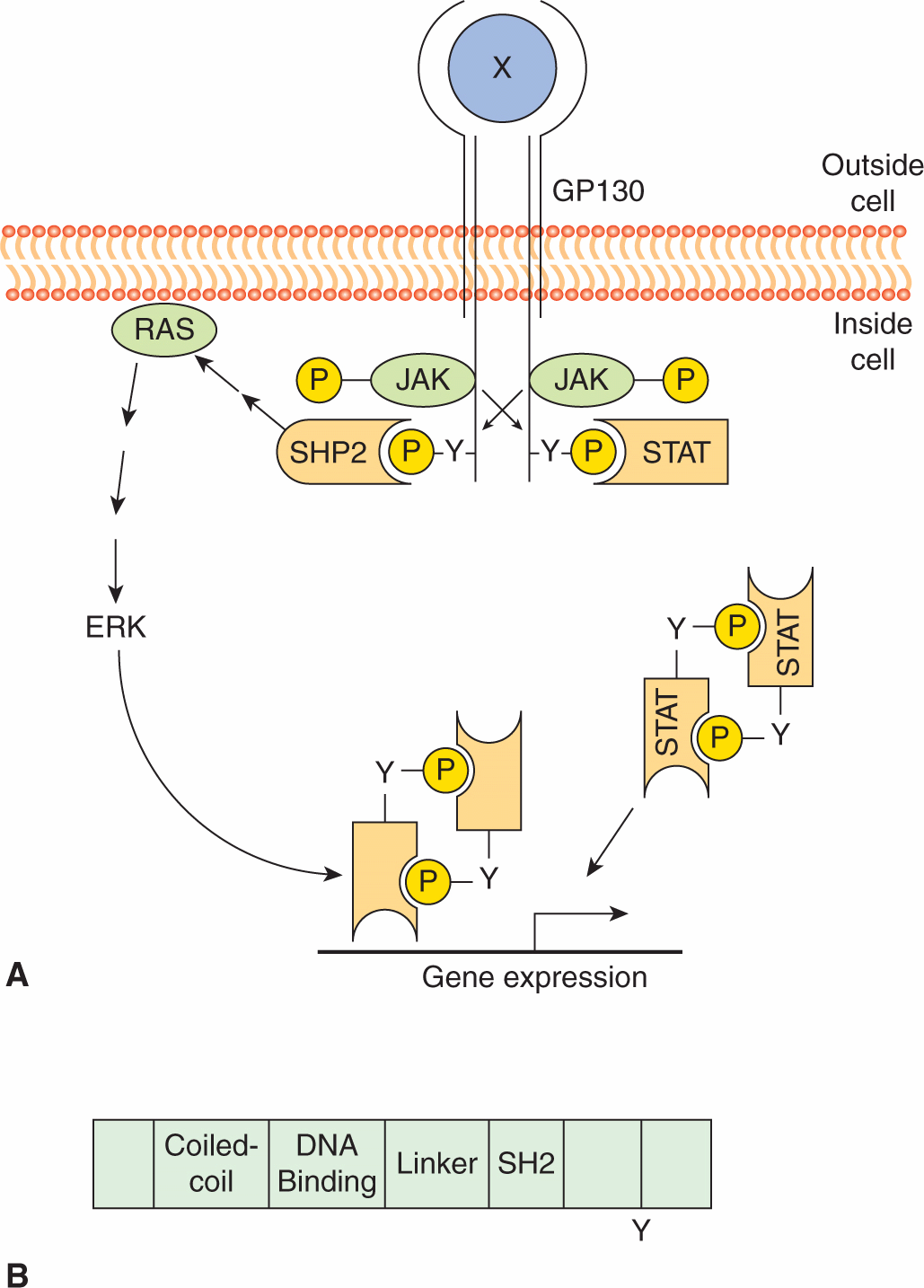
FIGURE 8–11 Cytokine receptor signaling. A) Cytokine (X) binding induces receptor dimerization resulting in JAK activation and phosphorylation of the GBP130 receptor subunit. STATs then bind the phosphorylated receptor, and are subsequently phosphorylated, inducing dimerization and transport to the nucleus where they activate downstream target genes. GBP130 phosphorylation also results in recruitment of tyrosine phosphatase SHP2, which in turn leads to RAS activation. B) Structure of the STATs. STATs contain a DNA binding domain, an SH2 domain, and a conserved tyrosine residue in the carboxy terminus.
Both gene translocations and activating point mutations in JAK2 have been identified in myeloproliferative disorders and leukemia. A common feature of the translocations is a protein product that fuses the kinase domain of JAK2 with a dimerization or oligomerization domain of the fusion partner leading to constitutive kinase activation (Jatiani et al, 2010). JAK2 point mutations, such as V617F, which causes substitution of valine at amino acid 617 for phenylalanine, are found frequently in myeloproliferative diseases. Valine 617 is within the pseudokinase domain and the V617F mutation is thought to disrupt the autoinhibitory interaction between the pseudokinase and catalytic kinase domains. Several JAK2 kinase inhibitors have been developed and assessed in clinical trials. For example, ruxolitinib is a JAK2 kinase inhibitor that has shown efficacy in reducing disease burden and improving quality of life through reduction of disease-related symptoms. However, JAK2 inhibitors do not improve bone marrow pathology or reduce the number of cells with the JAK2 mutant allele or cause disease remission. As with many other targeted agents, combination therapy may be required to achieve disease remission (Jatiani et al, 2010).
8.3.2 Integrin Signaling
Mammalian cells express cell adhesion molecules that mediate their attachment to the extracellular matrix (ECM) and/or their interaction with the same or different cell types (see Chap. 10, Sec. 10.2). Most adhesion molecules are transmembrane proteins, but some are anchored in the plasma membrane by an C-terminal glycophosphatidyl-inositol moiety. Interactions between cells and the ECM are essential for cell survival and cell proliferation, and can regulate differentiation. Loss of interactions between the cell and ECM results in induction of apoptosis in both epithelial and endothelial cells. However, a common property of malignant cells is that they continue to survive and proliferate in the absence of interactions with the ECM.
Integrins are transmembrane cell-surface receptors expressed in all cell types that serve as the primary physical link between the ECM and actin cytoskeleton, and they enable direct communication across the plasma membrane. Integrins recognize and bind to specific ECM ligands (such as collagen, laminin, and fibronectin) and transduce signals leading to the activation of intracellular signaling pathways that regulate cell migration, cell polarity, cell proliferation and survival. Integrins are comprised of membrane spanning α and β subunits that associate noncovalently to form a heterodimer on the cell surface. Receptor diversity and versatility in ligand binding is determined by the extracellular domains, and through the specific pairing of 9 β subunits and 16 α subunits (see Chap. 10, Fig. 10–3). The cytoplasmic domains of both α- and β-integrin subunits are conserved among vertebrate species and Drosophila, and serve as a binding platform for both actin cytoskeleton binding proteins, such as α-actinin, paxillin, tensin and talin, and intra-cellular signaling components, such as cytoplasmic TKs, Focal Adhesion KInase (FAK) and SRC families (Fig. 8–10) (Harburger and Calderwood, 2009).
The binding of integrins to ECM ligands induces integrin clustering and subsequent recruitment of actin filaments and signaling proteins to the integrin cytoplasmic domain (see Chap. 10, Fig. 10–3). These large transmembrane adhesion complexes composed of the ECM and intracellular signaling components are called focal adhesions (FAs). The dynamic formation and remodeling of FAs assures cell adhesion to the ECM in addition to the targeted localization of actin filaments and signaling components necessary for the establishment of cell polarity, directed cell migration, and maintenance of cell proliferation and survival (Scales and Parsons, 2011). In addition to providing a physical link between the ECM and the cytoskeleton, binding of integrins to their ligands elicits a variety of intracellular signaling events (Hynes, 2002; Liu et al, 2000; Martin et al, 2002). For example, integrin signaling regulates the formation of filopodia, lamellipodia, and stress fibers through the RHO family of small GTPases CDC42, RAC, and RHO. It can also stimulate tyrosine phosphorylation and subsequent activation of cellular proteins, including the FAK and SRC cytoplasmic TKs that regulate remodeling of FAs (Fig. 8–10). Furthermore, activation of the RAS family of GTPases by integrins is important for activation of serine-threonine kinases, such as ERK, PAK, and JNK, that regulate gene expression and cell-cycle progression (Scales and Parsons, 2011).
Both up- and downregulation of integrins have been observed in tumor progression, and changes observed in integrin expression appear to be tumor- and integrin-specific (Desgrosellier and Cheresh, 2010). Loss of integrin α2β1 is observed in some tumors while other integrins such as αvβ3, αvβ6, and α5β1 are upregulated in epithelial tumors and associated with disease progression, metastasis, and/or tumor-induced migration of vascular endothelial cells (see Chap. 10, Sec. 10.2.3). Monoclonal antibodies and RGD peptides that block integrin ligand binding or function have been investigated as potential cancer therapeutics, and promising preclinical studies have led to a Phase III trial of the integrin-blocking peptide cilengitide in glioblastoma (Desgrosellier and Cheresh, 2010).
8.4 DEVELOPMENTAL SIGNALING PATHWAYS
Although cell–cell signaling is a fundamental aspect of early development, only a few signaling pathways are required to generate the functional and morphological diversity of cell types and patterns found in most animals. These signaling networks include WNT, Hedgehog, transforming growth factor-β (TGF-β), Notch, JAK/STAT, and nuclear hormone pathways. Depending on the cellular context, these pathways activate specific target genes and produce a spectrum of cellular signals, resulting in effects on cellular physiology and embryonic development. Predictably, abnormal activation of these crucial pathways is observed in disease states such as cancer, and understanding these pathways is central to our knowledge of cancer etiology and to the development of targeted therapy.
8.4.1 WNT Signaling
The WNT proteins are signaling molecules necessary for development of a multicellular organism. They constitute a large family of secreted glycoproteins with 19 known human members that are highly conserved throughout evolution WNT proteins bind to the Frizzled (FZ) family of transmembrane receptors of which 10 members have been identified (Fig. 8–12) Together with their coreceptors, low-density lipoprotein receptor-related proteins LRP5 and LRP6, ligand-bound FZ receptors initiate signaling to downstream intracellular targets. The affinity of FZ receptors for specific ligands determines the activation of 3 alternate intracellular pathways: (a) the canonical (ie. major) WNT pathway, which leads to regulation of gene expression through β-catenin; (b) the WNT/planar cell polarity pathway, which activates cyto-skeleton reorganization through RHO and JNK; and (c) the WNT/Ca2+ pathway, which involves the activation of phospholipase C and protein kinase C (Gao and Chen, 2010).
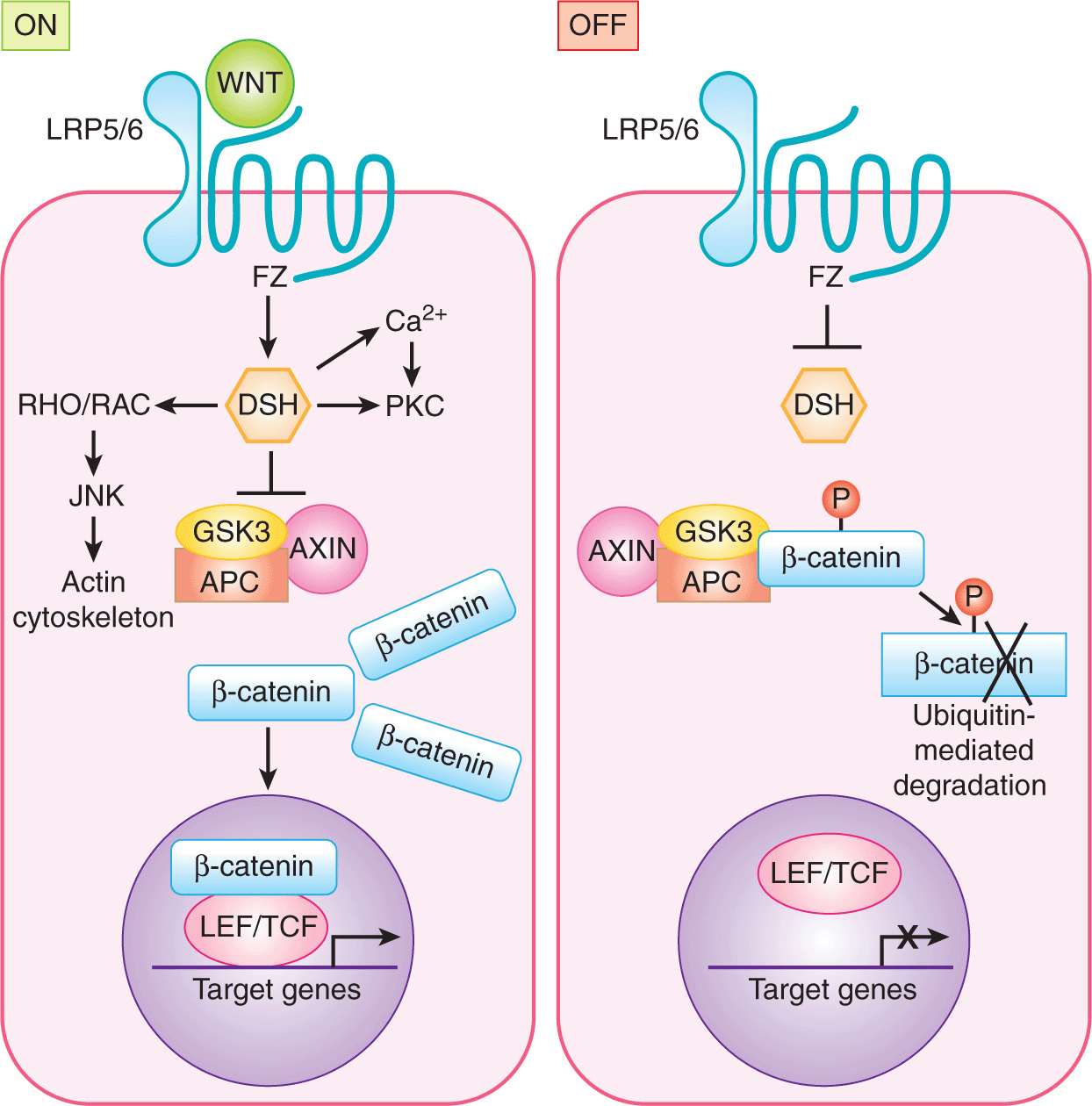
FIGURE 8–12 WNT signaling. ON: In the canonical WNT signaling pathway, WNT binding to Frizzled (FZ) LRP5/6 complex on the target cell results in recruitment of Dishevelled (DSH) to FZ. This interaction results in inactivation of the kinase glycogen synthase kinase 3 (GSK3), thereby stabilizing β-catenin. β-Catenin accumulates in the cytoplasm and shuttles to the nucleus where it functions as a cotranscriptional activator with LEF/TFC to regulate transcription of target genes. OFF: In the absence of WNT, β-catenin is phosphorylated and exists within a destruction complex (together with APC, GSK3, and AXIN) and is targeted for ubiquitin-dependent proteasomal degradation. The noncanonical WNT pathways include WNT/Ca2+ and WNT/Planar cell polarity pathways (see text).
The key mediator of canonical WNT signaling is β-catenin (Akiyama, 2000; Moon et al, 2002). In the absence of WNT, β-catenin is degraded through a series of phosphorylation and ubiquitination steps mediated by the “destruction complex” of proteins (Fig. 8-9; Kimelman and Xu, 2006). In the absence of β-catenin, T-cell factor (TCF) together with corepressors such as histone deacetylase (HDAC) and Groucho/transducin-like-enhancer of split (TLE) repress the transcription of WNT target genes. Upon WNT activation, the destruction complex is inactivated, allowing β-catenin accumulation and entry into the nucleus where it interacts with the lymphoid enhanced transcription factor (LEF) and the TCF family of transcription factors to regulate transcription of specific target genes. These transcription factors bind directly to DNA but are incapable of activating gene transcription independently of β-catenin. Known target genes include c-myc, cyclin D1, and metalloproteinase 7.
Constitutive activation of the canonical WNT pathway has been observed in many cancers including colorectal carcinoma (CRC). Adenomatous polyposis coli (APC) is a tumor suppressor that forms part of the destruction complex and is encoded by the gene responsible for the onset of familial adenomatous polyposis (FAP), an autosomal dominant, inherited disease that predisposes patients to multiple colorectal polyps and cancers. In this disease, mutations in the APC gene result in ineffective β-catenin degradation and constitutive activation of β-catenin/TCF target genes. Consistent with this inability, in 50% of cases of sporadic CRC, mutations that protect β-catenin from degradation have been observed (Morin et al, 1997). Activating mutations in β-catenin or in other components of the canonical WNT pathway are also observed in a wide variety of human malignancies including carcinoma of the lung, breast, cervix, stomach, pancreas, ovary, prostate, hepatocellular carcinoma (HCC), and medulloblastoma (Polakis, 2007).
Targeting the WNT pathway for cancer therapy has proven successful under experimental conditions. Small molecule antagonists of TCF/β-catenin interaction inhibit proliferation of CRC, HCC, and multiple myeloma cells (Lepourcelet et al, 2004; Sukhdeo et al, 2007; Wei et al, 2010). Furthermore, inhibition of the WNT target gene product COX-2 with nonsteroidal anti-inflammatory drug treatment of patients with FAP or a human FAP mouse model significantly reduces the number of intestinal polyps (Oshima et al, 1996; Steinbach et al, 2000).
Compounds that target the noncanonical WNT/Planar cell polarity or WNT/Ca2+ pathways are under development. Small molecule-mediated inhibition of RAC, a downstream effector of the WNT/PCP pathway, can suppress the growth and invasion of prostate cancer cells and leukemia cells under experimental conditions (Gao et al, 2004; Wei et al, 2008). Inhibition of PKC (protein kinase C), a WNT/Ca2+ pathway effector has demonstrated limited success in a number of malignancies including melanoma, non-Hodgkin lymphoma, and ovarian cancer (Swannie and Kaye, 2002). FOXY-5, an agonist of the noncanonical WNT tumor suppressor, WNT5a, can eradicate breast cancer in a mouse model (Safholm et al, 2008). In summary, both canonical and noncanonical WNT signaling pathways are potential targets for cancer therapy, but no specific modulator of WNT signaling has yet progressed into clinical trials.
8.4.2 Notch Signaling
The Notch signaling cascade is highly conserved and plays a crucial role in stem cell self-renewal (see Chap. 13, Sec. 13.4.1), cell fate determination, epithelial cell polarity/adhesion, cell division, and apoptosis (see Chap. 9, Sec. 9.4.2) in organisms from nematodes to vertebrates. Notch is a ligand-activated cell-surface receptor initially identified in Drosophila, and subsequently identified in Caenorhabditis elegans and vertebrates. Mammals possess 4 Notch proteins (NOTCH1-4) that function as receptors for 5 Notch ligands (DELTA-LIKE1, -3, -4 and JAGGED1, -2) (Fig. 8–13). Glycosylation by Fringe glycosyltransferases (LUNATIC, MANIC and RADICAL FRINGE) modifies the specificity of ligand-receptor interactions (Callahan and Egan, 2004). The Notch receptor is a large, single-pass, transmembrane protein that contains a number of conserved protein–protein interaction motifs within its 3 domains (Fig. 8–13). The large extracellular domain contains a number of EGF-like repeats involved in ligand binding, and 3 cysteine-rich Lin12/Notch repeat (LNR) regions thought to play an inhibitory role in receptor activation. This region is followed by a single transmembrane domain, and an intracellular domain composed of a RAM23 site that interacts with proteins from the CSL (CBF1/SU[H]/LAG1) family of transcription factors, 6 tandem ankyrin repeats involved in mediating protein–protein interactions with regulators of the receptor, and a proline, glutamine, serine, and threonine-rich (PEST) sequence associated with high rates of protein turnover.
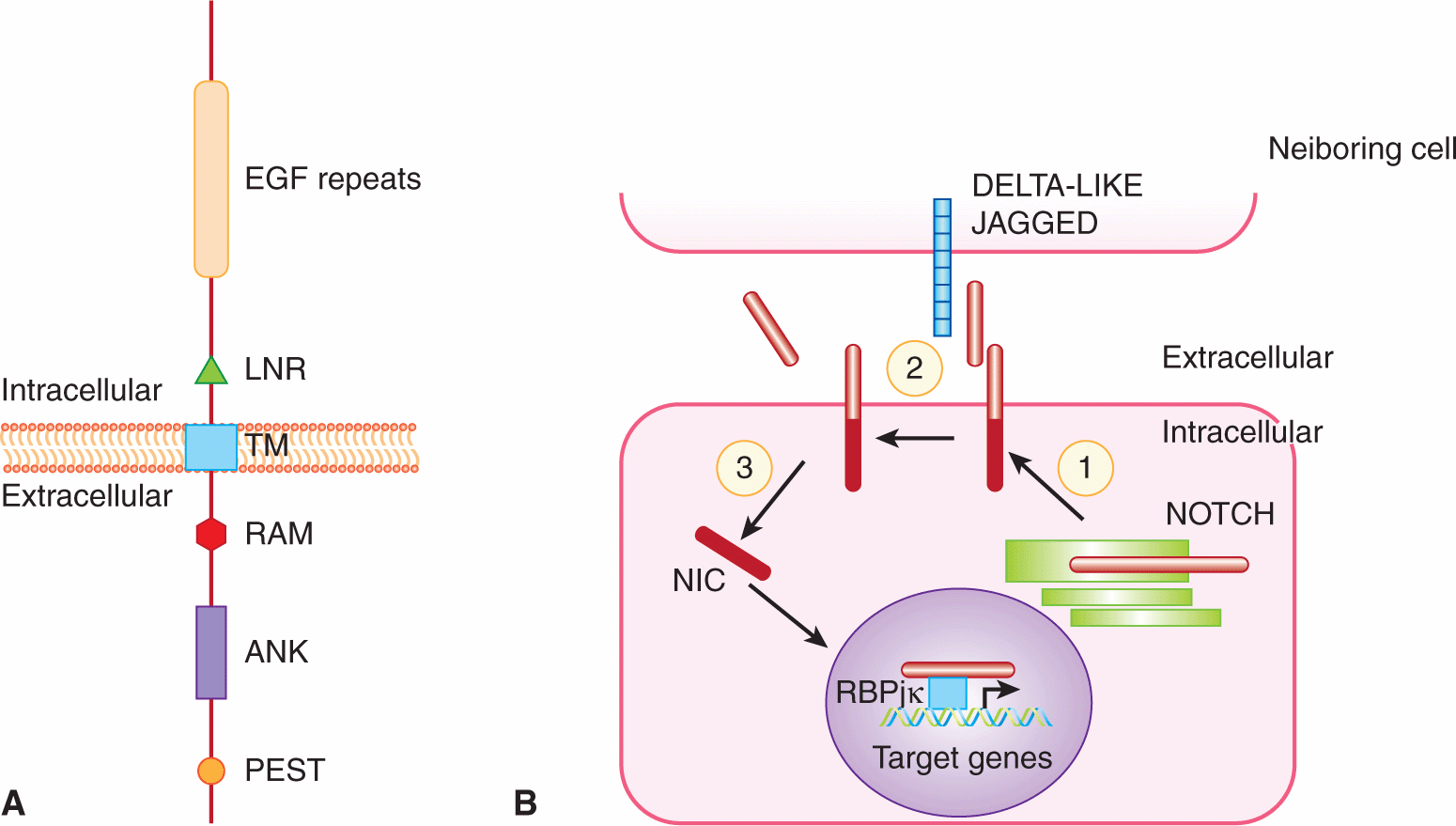
FIGURE 8–13 Notch receptor signaling. A) Schematic of the NOTCH receptor. EGF, Epidermal growth factor-like repeats; LNR, Lin12/Notch repeat region; TM, transmembrane domain; RAM, RAM23 domain; ANK, ankyrin repeats; PEST, proline, glutamate, serine, threonine-rich region. B) Model of Notch pathway. (1) NOTCH is first processed in the trans-Golgi network into a heterodimer which is found on the cell surface. (2) Ligand (DELTA-LIKE, JAGGED) presented on neighboring cells causes a second cleavage event that releases the extracellular domain of NOTCH. (3) This triggers a final cleavage event mediated by the γ-secretase complex, releasing the active intracellular domain of NOTCH (NIC). NIC translocates to the nucleus where it functions as a cotransactivator with the CSL family of transcription factors (such as RBPjk) to regulate target genes.
Notch signaling involves complex cleavage events that occur during receptor maturation and transmission of the Notch signal. During its maturation, NOTCH is first processed into 2 distinct fragments that interact to form a heterodimer on the cell surface (see Fig. 8–13B). In this heterodimeric form, NOTCH is able to bind transmembrane ligands presented on neighboring cells. Notch ligand-receptor interaction leads to 2 activating proteolytic cleavages of the receptor. The first cleavage event, mediated by metalloproteases of the ADAM (a disintegrin and metalloproteinase) family, releases the Notch extracellular domain. The second cleavage, executed by a presenilin-protease (γ-secretase) complex, releases the cytoplasmic domain fragment of intracellular NOTCH (NIC) from the plasma membrane (Iwatsubo, 2004; Xia and Wolfe, 2003). NIC enters the nucleus and modulates the expression of target genes predominantly by converting the CSL repressor to an activator of transcription (Honjo, 1996). Downstream targets of CBF-1 include members of the HES and HEY families, which encode basic helix-loop-helix transcriptional regulatory proteins (see Sec. 8.2.6; Iso et al, 2003).
Notch activation has been strongly linked to human cancers, most notably T-cell acute lymphoblastic leukemia (T-ALL). Here, a recurrent t(7;9)(q34;q34.3) chromosomal translocation, resulting in a dominantly active NOTCH1 receptor, was identified in a small subset of tumors (Ellisen et al, 1991). Weng et al (2004) discovered that more than 50% of T-ALLs harbored gain-of-function mutations within the NOTCH1 receptor.
Aberrant Notch signaling can also contribute to the development of solid malignancies such as breast cancer. The Notch4 locus was identified as a common proviral integration site in MMTV-induced mammary adenocarcinomas in mice, leading to overexpression of NIC protein (Gallahan and Callahan, 1987). In human breast cancer, high-level expression of JAGGED1 and/or NOTCH1 correlates with and is an independent prognostic indicator of poor outcome (Reedijk et al, 2005). Activated Notch signaling and upregulation of genes that are required for tumor growth have been observed in breast cancer cell lines (Stylianou et al, 2006). Notch affects multiple cellular processes and aberrant Notch activity influences breast cancer progression through the maintenance of tumor-initiating cells and by promoting proliferation, motility, and survival of cancer cells. Aberrant Notch signaling has also been identified in several other solid malignancies, including prostate cancers and head and neck squamous cell carcinomas. Thus, aberrant Notch signaling contributes to the progression of a wide spectrum of human cancers.
Notch ligands or receptors may be inhibited by monoclonal antibodies, RNA interference, soluble ligands, or receptor decoys. Inhibition of enzymes involved in glycosylation or cleavage of receptors, such as γ-secretase inhibitors (GSIs) or ADAM inhibitors are also potential approaches to target Notch therapeutically. Among Notch pathway inhibitors, GSIs have the most immediate therapeutic potential. Potent GSIs have been available for over a decade and were developed originally to inhibit the γ-secretase complex that plays a role in plaque formation in Alzheimer disease. The discovery of γ-secretase-dependent NOTCH1 mutations in T-ALL accelerated the use of GSIs in preclinical studies, where induction of apoptosis and reduced cell proliferation was demonstrated in several human cancer models, including Notch-activated breast cancer cells cultured as a monolayer (O’Neill et al, 2007) or as tumor spheres (Farnie et al, 2007). GSI treatment also blocks Notch signaling and growth of human breast cancer xenografts (Rizzo et al, 2008) and tumors in a HER-2 transgenic breast cancer mouse model (Watters et al, 2009). Several Phase I and II clinical trials with GSIs are underway.
8.4.3 Hedgehog Signaling
Hedgehog (HH) proteins act as key mediators of fundamental processes in embryonic development, and serve broad roles in the proliferation, migration, and differentiation of target cells, and in the maintenance of stem cell populations (Merchant and Matsui, 2010). HH signaling is essential to the growth, patterning, and morphogenesis of many different tissues and organs including the skin, brain, gut, lung, and bone, and HH proteins play a role in hematopoiesis (Cohen, 2003; Ingham and McMahon, 2001; Nybakken and Perrimon, 2002). The HH signaling pathway is highly conserved throughout evolution, and much of what is known about signaling in vertebrates has been inferred from studies in Drosophila. In humans there are 3 homologs of the Drosophila Hh gene; sonic hedgehog (Shh), desert hedgehog (Dhh), and Indian hedgehog (Ihh). The Hh gene products encode ligands that signal through a membrane–receptor complex including the Patched (PTC1 and PTC2) and Smoothened (SMO) receptors, which together form a molecular switch controlling activation of downstream target genes (Fig. 8–14). In the absence of ligand, PTC inhibits SMO accumulation and activation (Rohatgi et al, 2007). The interaction between HH and PTC releases SMO (Corbit et al, 2005; Rohatgi et al, 2007). Activated SMO allows expression and/or proteolytic processing of 3 zinc-finger transcription factors (see Sec. 8.2.6; GLI1, -2, and -3) and ultimate transcription of HH target genes through direct interaction with the consensus binding sequence of bases in DNA 5′-tgggtggta-3′. Several regulators of HH signaling have been identified, including molecules that modify the ligand, such as Hedgehog interacting protein (HIP) and Hedgehog acyltransferase (HHAT), and proteins that function downstream of SMO, including the serine/threonine protein kinase Fused (FU) and the Suppressor of Fused (SU[FU]) (reviewed in Merchant and Matsui, 2010).
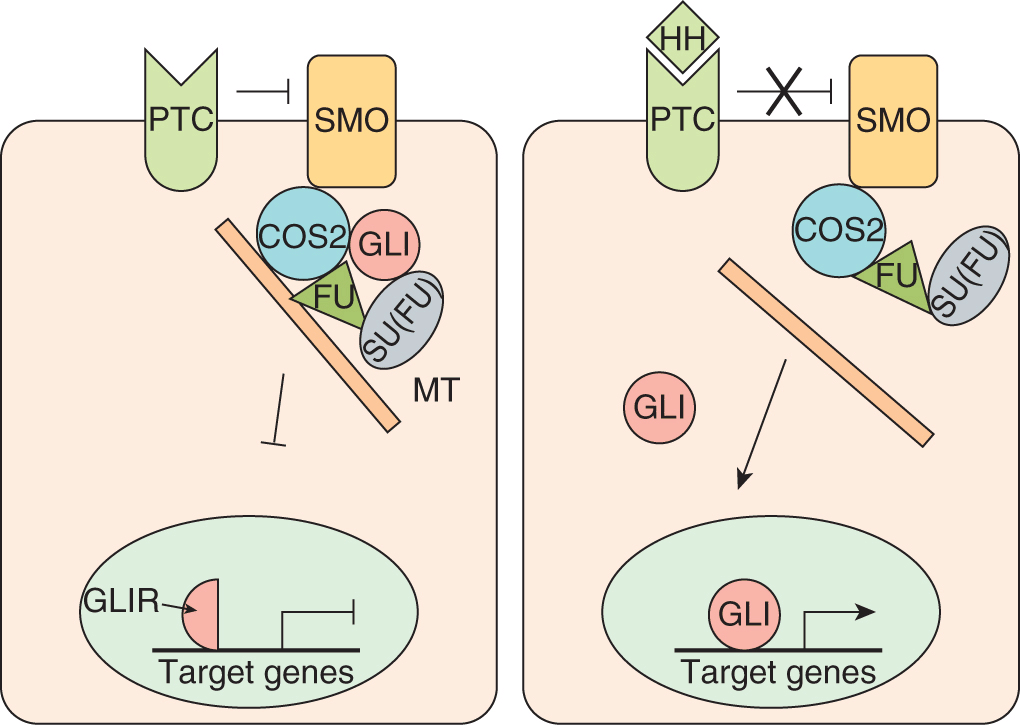
FIGURE 8–14 Hedgehog signaling. In the absence of HH, PTC silences SMO, the key signal transducer of the HH pathway. Binding of HH to PTC prevents SMO inhibition and results in nuclear translocation of activated Gli. Release of SMO inhibition results in nuclear translocation of activated GLI transcription factors and HH target gene expression. GLI is found in a complex with Fused (FU), Costal2 (COS2), and Suppressor of Fused (SU[FU]) bound to microtubules (MT). Upon activation of SMO this complex is disassembled, releasing GLI and allowing nuclear translocation and upregulation of downstream target genes. Target genes include WNT, bone morphogenetic protein (BMP), and Ptc itself.
Given its critical role in development, disturbances in HH signaling can result in disease states. Inactivation of the HH signaling pathway during development results in an abnormality in which the embryonic forebrain fails to develop into 2 hemispheres, whereas abnormal activation of this pathway has been implicated in the development of malignancy. HH pathway activation in cancer may occur through different mechanisms, including activating mutations within components of the signaling pathway. For example, Ptc1 loss-of-function or Smo gain-of-function mutations have been observed in sporadic basal cell carcinoma (BCC) and medulloblastoma (reviewed in Pasca di Magliano and Hebrok, 2003). Consistent with these observations, Gorlin (basal cell nevus) syndrome, which results from Ptc1 mutation, leads to a predisposition to BCC, medulloblastoma, and rhabdomyosarcoma (Hahn et al, 1996; Johnson et al, 1996). These findings have been recapitulated in mouse models of SMO activation or PTC inactivation, where animals frequently develop BCC or medulloblastoma (Pasca di Magliano and Hebrok, 2003). Gli activation can also result in malignancy. This propensity may occur as a result of K-ras activating mutations that increase GLI transcriptional activity or through mutations within the Gli genes themselves; both mechanisms of HH activation have been observed in carcinoma of the pancreas (Ji et al, 2007; Jones et al, 2008). Mutations in Su(Fu), have been identified in patients with medulloblastomas (Taylor et al, 2002). HH signaling in malignancy can also be induced by pathways that are commonly activated in human cancer, such as PI3K/AKT and MEK (see Sec. 8.2.4; Stecca et al, 2007).
Aberrant HH activation may also occur through ligand-mediated mechanisms in either autocrine or paracrine settings (reviewed in Merchant and Matsui, 2010). In multiple malignancies, including small cell lung cancer (SCLC), pancreatic adenocarcinoma, colon cancer, prostate cancer, glioblastoma, and malignant melanoma, tumor cells have been shown to synthesize HH ligand to which they also respond. In B-cell lymphoma and multiple myeloma, stromal production of HH ligand in the spleen, lymph nodes, and bone marrow supports the growth of tumor cells. Conversely, tumor production of HH ligand can stimulate HH signaling in stromal cells of the tumor microenvironment, resulting in the expression of HH target genes in those cells to increase the stromal content of tumors. Indeed, inhibition of HH signaling in tumor stroma in a mouse model of pancreatic carcinoma has been reported to improve delivery of cytotoxic chemotherapy (Olive et al, 2009).
A role for HH signaling in the maintenance of tumor initiating cells (TICs; see Chap. 13, Sec. 13.4) has been reported for multiple tumor types, including breast and pancreatic cancer, glioblastoma, multiple myeloma, and chronic myeloid leukemia (Merchant and Matsui, 2010). Analogous to normal tissue stem cells, TICs are defined by their capacity to self-renew and to regenerate tumors containing differentiated progeny identical to the original tumor.
Also suggestive of a putative role in TICs is emerging evidence that implicates HH signaling in tumor invasion and metastases (Feldmann et al, 2007). In primary colon cancer, HH signaling is upregulated in the TIC compartment and is accompanied by elevated expression of Snail, a protein involved in epithelial-to-mesenchymal transition (EMT) and metastasis (Varnat et al, 2009). Growth and metastases of primary colon cancer in a xenograft model requires active HH signaling and induction of EMT.
Numerous HH antagonists are undergoing clinical testing in a wide range of malignancies including BCC. An early encouraging report of the Smo antagonist GDC-0449, demonstrated a 55% clinical response rate, including 2 complete remissions in 33 patients with advanced BCC (Von Hoff et al, 2009). While these results are promising, results from ongoing Phase I and II clinical trials in SCLC, glioblastoma, pancreatic adenocarcinoma, colon cancer, prostate cancer, and other malignancies are pending.
8.4.4 Signal Transduction by the Transforming Growth Factor-β Superfamily
Members of the TGF-β superfamily regulate a number of developmental and homeostatic processes, such as cell proliferation, differentiation, apoptosis, cell adhesion, and migration. They constitute a highly conserved family of proteins with at least 30 vertebrate members and over a dozen structurally and functionally related proteins found in invertebrates, such as C. elegans and Drosophila. There are 2 general branches of this superfamily, including a TGF-β/activin/nodal branch and a bone morphogenic protein (BMP) branch whose members have diverse, but often complementary, effects. Some members are widely expressed during embryogenesis and in adult tissues, whereas others are expressed in only a few cell types and for restricted periods during development.
TGF-β, the prototypic member of this superfamily is a secreted growth factor synthesized as an inactive precursor and is proteolytically processed into a mature secreted ligand. Upon dimerization, TGF-β becomes biologically active and binds to a cell-surface receptor complex consisting of 2 distinct single-pass transmembrane proteins known as the type I and type II receptors, both of which contain an intracellular serine-threonine kinase domain (Fig. 8–15; Attisano and Wrana, 2002; Dennler et al, 2002; Moustakas et al, 2001). Ligand binding induces association of the type I and type II receptors into a heterotetrameric complex. This association leads to unidirectional phosphorylation and subsequent activation of the kinase domain of the type I receptor by the type II receptor. The activated type I receptor then signals to the SMAD (for Sma and Mad proteins from C. elegans and Drosophila, respectively) family of intracellular mediators, which function to carry the signal from the cell surface directly to the nucleus. There are 3 distinct classes of SMADs: receptor-regulated or R-SMADs (SMADs 1, 2, 3, 5, and 8), common mediator or Co-SMAD4, and inhibitory SMADs 6 and 7 (Fig. 8–15). The activated type I receptor directly phosphorylates and activates the R-SMADs leading to interaction with Co-SMAD4. The inhibitory SMADs counteract the effects of R-SMADs and antagonize TGF-β signaling. An additional level of regulation of TGF-β signaling occurs with SMURF1 and SMURF2. These proteins are E3 ubiquitin-protein ligases that regulate SMAD levels. SMURFs orchestrate ubiquitin transfer to the R-SMADs causing their ubiquitination and subsequent proteasomal degradation (see Fig. 8–9).
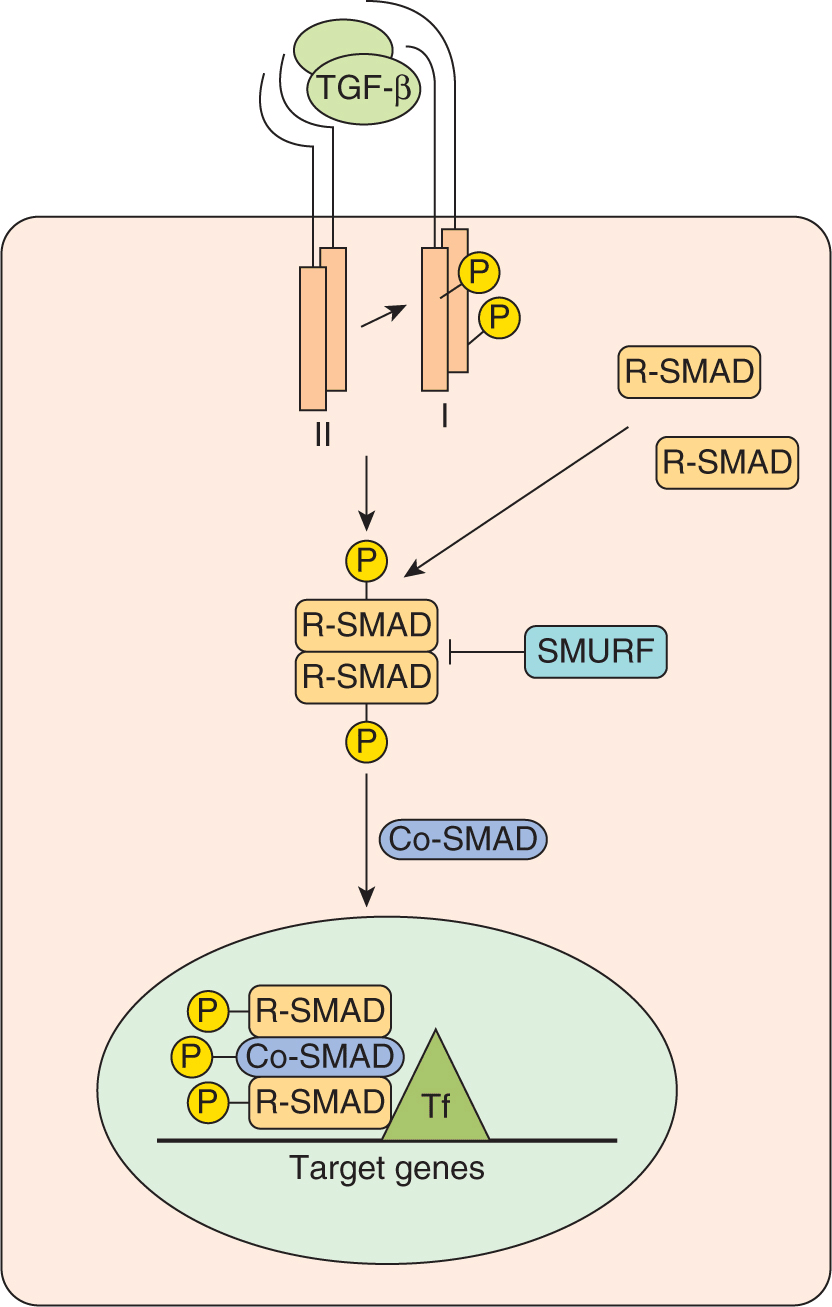
FIGURE 8–15 TGF-β signaling. TGF-β ligand binding induces the association of type II and type I receptor heterodimers into a heterotetrameric complex. This results in phosphorylation and subsequent activation of the type I receptor that then phosphorylates a member of the R-SMAD class of proteins (SMADs 1, 2, 3, 5, or 8). Phosphorylated R-SMADs interact with the Co-SMAD, SMAD4, and this complex then accumulates in the nucleus. SMURFs are E3 ubiquitin-protein ligases that orchestrate ubiquitin transfer to the R-SMADs causing their ubiquitination and subsequent proteasomal degradation. In the nucleus, the activated SMAD complex associates with DNA binding cofactors, coactivators and corepressors that regulate transcription of target genes. Tf, Transcript ion factors.
The biological output of TGF-β signaling appears to be almost entirely determined by the type I receptor. In vertebrates there are 7 distinct type I receptors that interact with 1 of 5 type II receptors. The signal from the type I receptor is funneled through 1 of 2 groups of SMAD proteins. Specific R-SMADs recognize different DNA binding proteins and regulate distinct target genes, thereby generating diverse biological responses. For example, phosphorylation and activation of R-SMADs SMAD2 and SMAD3 transduce a TGF-β-like signal, whereas activation of R-SMADs SMAD1, SMAD5, and SMAD8 transduce signals initiated by bone morphological proteins. There is also evidence that TGF-β can signal through SMAD independent processes through the activation of RHOA, RAS, and TGF-β activated kinase I (Tak1) (Massague et al, 2000).
Mutations in the TGF-β family of ligands are responsible for a variety of human diseases, including human cancer, hereditary chondrodysplasias, and pulmonary hypertension (de Caestecker et al, 2000; Massague et al, 2000). TGF-β signaling plays an important role in cancer progression by functioning as both an antiproliferative factor and as a tumor promoter. For example, in colon cancer, TGF-β can switch from an inhibitor of primary tumor growth to a stimulator of proliferation in metastatic cells (Lampropoulos et al, 2012). Immunohistochemical staining intensity of TGF-β correlates significantly as an independent marker of colon cancer progression to metastases. Mutations in the TGF-β receptors have been identified in several human cancers with a recent meta-analysis demonstrating that the TGF-β type I 6A polymorphic allele (containing a deletion of 3 alanine residues from the N-terminus) is associated with increased cancer risk (Zhang et al, 2005). In addition, mutations in SMAD2 are found in colorectal and lung cancers, and SMAD4 mutations are found in a large number of colorectal, pancreatic, and lung cancers (Lampropoulos et al, 2012). These point mutations in the SMAD proteins lead to loss of phosphorylation of SMAD2, for example, and subsequent loss of association with SMAD4.
Agents that target TGF-β signaling are being developed, including TGF-β neutralizing antibodies, soluble TGF-βR:Fc fusion proteins, antisense oligonucleotides and inhibitors of TGF-β receptors. Some agents have reached clinical trial, including AP-12009, an antisense oligonucleotide targeting TGF-β2 messenger RNA that led to remission in some patients (Hau et al, 2007) and is in Phase III clinical trial for aggressive brain tumors. Because the TGF-β pathway has a dual role as both tumor suppressor and promoter, the clinical delivery of agents that modify TGF-β signaling must be carried out thoughtfully and their impact interpreted carefully.
Related posts:
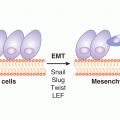
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


