Cell Proliferation and Death
9.1 INTRODUCTION
Our cells are continuously proliferating and dying. Our development from a single-celled egg into adults with approximately 1014 cells requires intense cell proliferation. But our development also requires cell death; for instance, to prune excess neurons in the brain and to sculpt the fingers. As adults, most of our organs exist in a dynamic steady state, being constantly renewed by cell proliferation and death. For example, our bodies generate and destroy more than a million blood and intestinal cells every second. In the extreme, cells live for only a few days before dying, as is the case for neutrophils and the cells that line the small intestine. In the midst of this continual and profuse cell renewal lies the constant threat of cancer. Cancerous cells invariably contain alterations to genes encoding regulators of cell proliferation and cell death and are generally thought to arise from actively proliferating cell types.
This chapter discusses the molecular control of the cell cycle and of the processes that lead to cell death, many of which are modified in the processes of malignant transformation and tumor progression. Chapter 12 discusses the growth of tumors and the patterns of cell proliferation and cell death that influence tumor growth, together with the tumor microenvironment and metabolism with which they are closely linked. Chapter 13 discusses the properties of tumor stem cells with high proliferative potential.
9.2 MOLECULAR CONTROL OF CELL PROLIFERATION
Cell proliferation is perhaps best viewed as a combination of two distinct processes: the cell cycle, which replicates and segregates the genome, and cell growth, which doubles all the other components of the cell. The cell cycle and cell growth are intertwined in most normal and cancerous cells, but the two processes can be uncoupled, both in the laboratory and as part of normal development.
9.2.1 The Mammalian Cell Cycle
The cell cycle is partitioned into 4 phases: G1, S, G2, and M. This organization reflects the 2 primary goals of the cell cycle: to replicate the genome of the mother cell during DNA synthesis or S-phase, and to segregate the replicated genome into 2 daughter cells during mitosis or M-phase (Fig. 9–1). Two gap phases (G1, G2) separate these fundamental events. The combined G1, S, and G2-phases are frequently referred to as interphase. When cells cease proliferating because of insufficient nutrients, lack of growth factors, or upon differentiation, they exit the cell cycle from G1-phase and enter a quiescent state called G0. Most cells in the body are in the G0 state. If cells in G0 are instructed to start proliferating, they must transition back into G1-phase before starting another cell cycle.
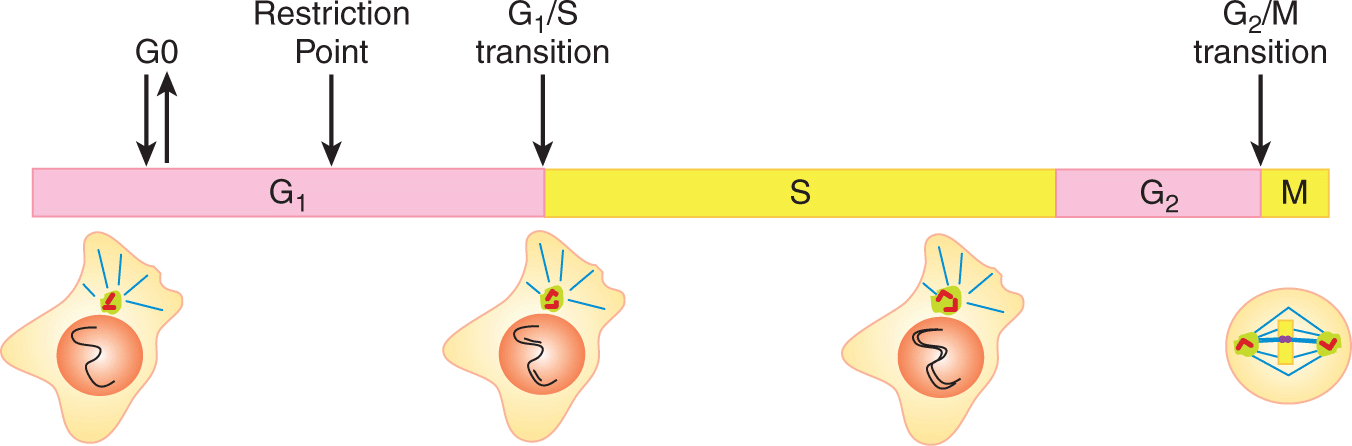
FIGURE 9–1 The key events of the cell cycle. In cells, the nucleus (orange) contains chromosomes (black). The centrosomes (green) are centred around centrioles (red). Most microtubules (blue) project from the centrosome. Chromosome and centrosome duplication begin at the start of S-phase. During mitosis, chromosomes (yellow) condense and the 2 sister chromatids become apparent, remaining joined at the centromeres, which is also the location of the kinetochores (purple) that bind the sister chromatids to bundles of microtubules from opposing poles. Cell structures are not drawn to scale.
9.2.1.1 The Genome Is Duplicated During S-Phase Replicating the genome is the first of the cell cycle’s two primary objectives. Each of the more than 6 billion base pairs in the DNA of a human cell must be replicated and replicated only once. To replicate so many base pairs, DNA synthesis is initiated at thousands of replication origins scattered throughout the genome (Fig. 9–2A,B). At each replication origin, double-stranded DNA (dsDNA) is unwound into 2 single stands and large protein complexes containing DNA polymerase load onto the single-stranded DNA (ssDNA) (Fig. 9–2C). As they start to replicate the ssDNA, these protein complexes progress away from the origin, unwinding the dsDNA in front of them and leaving 2 copies of dsDNA in their wake, thereby creating a structure called a replication fork (Fig. 9–2C). When replication forks progressing away from neighboring origins of replication collide, replication stops, the DNA replication complexes are removed, and the DNA strands are ligated together (Fig. 9–2A).
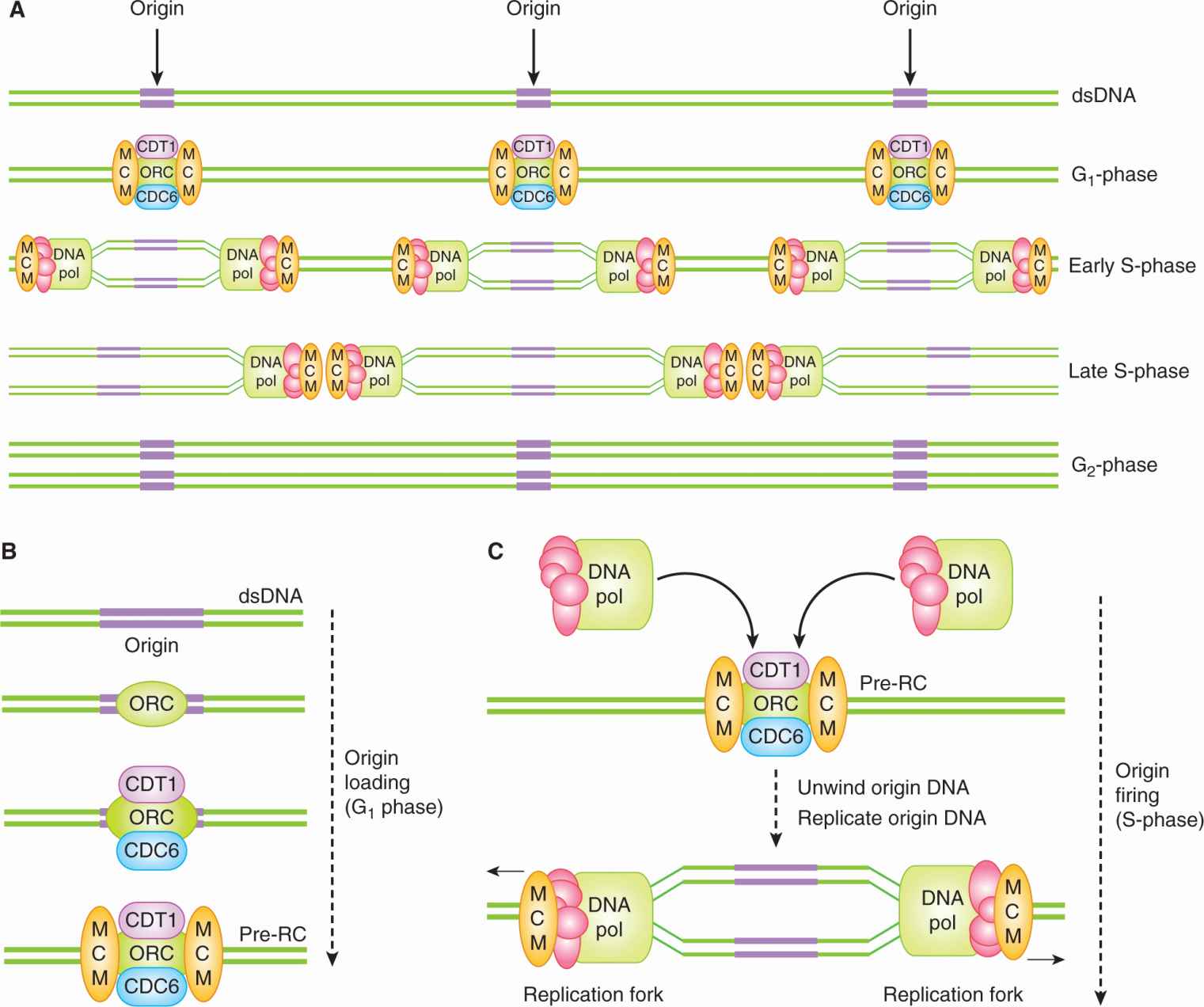
FIGURE 9–2 The loading and firing of DNA replication origins during the cell cycle. A, B) During G1 phase, prereplication complexes (pre-RCs) composed of ORC, CDT1, CDC6 and MCM proteins assemble on stretches of double-stranded DNA (dsDNA) termed replication origins (origins). ORC, Origin-recognition complex; MCM, minichromosome maintenance complex. A, C) In S-phase, replication proteins, including DNA polymerase (DNA pol), are recruited by the pre-RC, leading to the unwinding of the origin DNA and the exposure of single-stranded DNA (ssDNA). The 2 exposed stands of ssDNA act as the templates for DNA replication, leading to the formation of 2 copies of dsDNA. Replication proceeds bidirectionally away from the origin, as 2 replication forks unwind DNA and then replicate the exposed ssDNA.
Cells ensure that each stretch of interorigin DNA is replicated only once by allowing each DNA replication origin to initiate or “fire” only once per cell cycle. Origins can only initiate synthesis once each cell cycle because of a temporal separation between the formation of the prereplicative complex (pre-RC) on the origin and the initiation of replication at the origin (origin firing) (Arias and Walter, 2007). Pre-RCs are protein complexes containing DNA-binding proteins that assemble on origins at the end of the previous mitosis (see Fig. 9–2B). Once an origin is bound by a pre-RC, it is said to be “licensed” for replication, but these licensed origins remain inert throughout G1-phase. It is only starting in S-phase that licensed replication origins “fire,” when the aforementioned replication complexes containing DNA polymerases are recruited to the pre-RC and DNA synthesis begins at the origin (see Fig. 9–2C). Pre-RC complexes cannot reassemble in S-phase, nor throughout the subsequent G2 and early M phases. This strict separation between times during which pre-RCs can assemble and hence license the origins (G1-phase) and during which the licensed origins can fire (S-phase), limits each origin to a single firing event and prevents rereplication of the DNA.
9.2.1.2 The Duplicated Genome Is Segregated into Two Daughter Cells During Mitosis Segregating the replicated genomes into two daughter cells is the cell cycle’s other primary objective. Chromosome segregation is powered and organized by microtubules. Microtubules and associated proteins form the mitotic spindle, a complex cellular apparatus that pulls apart the replicated chromosomes and then drags the separated sister chromatids to opposite ends of the mother cell.
Assembly of the mitotic spindle is complicated, but is facilitated by centrosomes, organelles that sit at either spindle pole and generally act to organize microtubules. Centrosomes are small (~1 μm) and consist of an electron-dense pericentriolar material of uncertain organization centered on 2 compact (200 × 500 nm), barrel-shaped cylinders of microtubules called centrioles. Normal cells in G1-phase contain a single, centrally located centrosome with 2 separated centrioles (see Fig. 9–1). This single centrosome must replicate itself during the cell cycle. Centrosome replication begins in early S-phase when a daughter centriole grows orthogonally off the surface of each of the 2 original centrioles, leading to the formation of 2 proximal, but distinct, centrosomes, each centered on 2 orthogonally connected centrioles. Upon entering mitosis, the 2 centrosomes separate and move to opposite sides of the cell, with the spindle forming between them. Near the end of mitosis, the centrioles within each centrosome disengage, losing their orthogonal juxtaposition and drifting slightly apart to return to the G1-phase configuration.
At the start of mitosis, the replicated chromosomes (46 in humans) are comprised of 2 sister chromatids that are glued together along their entire length by cohesive protein complexes. Although it involves multiple parallel processes that progress in a continuous fashion, mitosis continues to be described as a series of discrete phases, defined by the landmark morphological events observed under the microscope by early cell biologists (Fig. 9–3). Prophase begins with chromosome condensation in the nucleus. As long linear molecules of DNA, chromosomes must be compacted approximately 10,000-fold in order to be cleanly separated from one another and to be moved around the cell (Morgan, 2007). In the cytoplasm, the 2 centrosomes separate from one another and the microtubule spindle begins to elaborate between them, pushing the centrosomes to opposite sides of the cell. At the beginning of prometaphase, the nuclear envelope breaks down, allowing the microtubule spindle to physically interact with the chromosomes (Guttinger et al, 2009). At this point, the 2 sister chromatids that make up each chromosome have condensed into distinct rods. During prometaphase, the sister chromatids lose most, but not all, of their length-wise cohesion but remain tightly juxtaposed at their centromeres (Peters et al, 2008). The centromere is a single, long (0.2 to 7 megabases) sequence of repetitive DNA encoded in each sister chromatid which forms a platform for the kinetochore, a large protein complex that will physically link microtubules with the sister chromatid (see Fig. 9–1). During prometaphase, each kinetochore is bound by a bundle of 20 to 25 microtubules (Walczak et al, 2010). For each sister chromatid pair, the 2 bundles emanate from opposite spindle poles. The bundles apply pulling forces on each centromere toward their respective spindle pole, but the sister chromatids remain tightly cohered at their centromeres. Under the influence of these counterbalanced pulling forces, as well as other spindle forces, each of the 46 pairs of sister chromatids becomes bi-orientated in the center of the spindle, aligned in a plane called the metaphase plate (Dumont and Mitchison, 2009). The cell is now in metaphase, but progresses into anaphase when an abrupt and total loss of sister chromatid cohesion occurs, allowing the opposing microtubule bundles to pull the sister chromatids apart and drag them to opposite spindle poles. The spindle itself then elongates, driving the divided genomes to opposite ends of the mother cell. During the subsequent telophase, the events of early mitosis are reversed, as the chromosomes decondense, the nuclear envelopes assemble, and the spindle is taken apart. The process of cytokinesis, which splits the mother cell into 2 daughter cells, is initiated in late anaphase and is completed when both daughter cells are in early G1-phase.
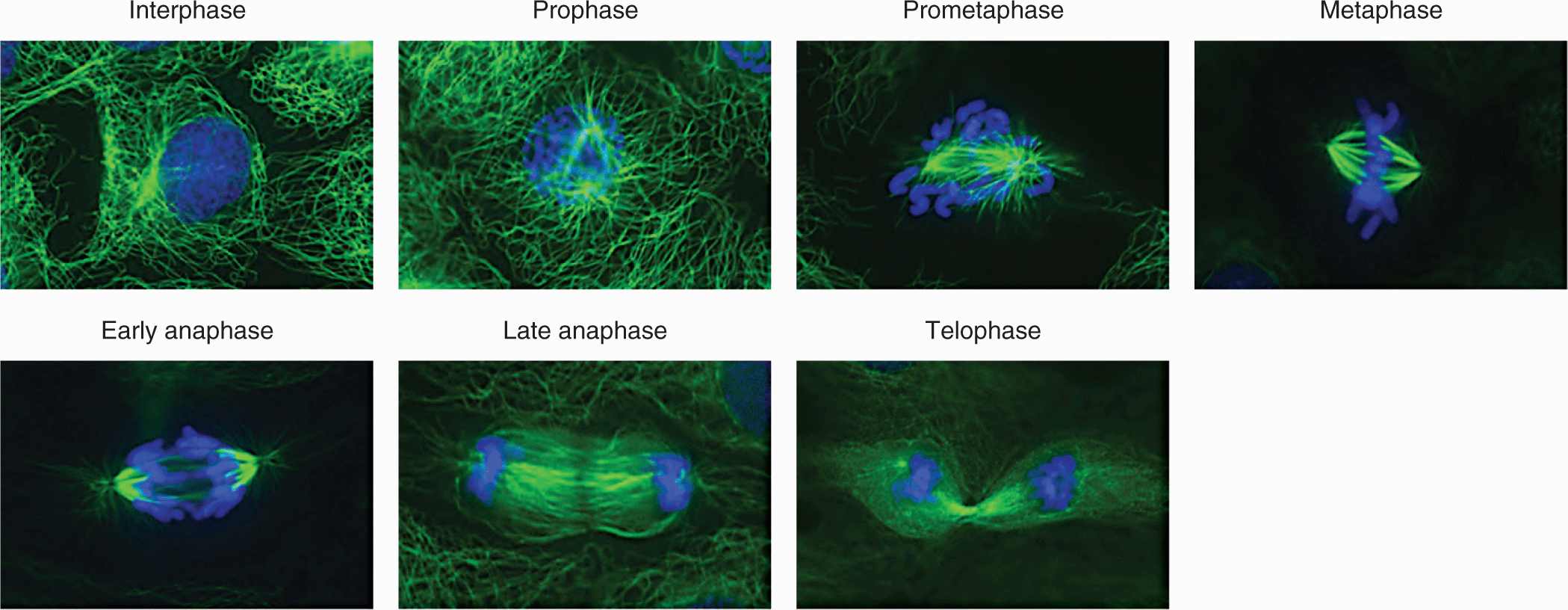
FIGURE 9–3 The phases of mitosis. Microtubules stained green, DNA stained blue. The events of each phase are described in the text. Note the microtubule-rich midzone between the segregated chromosomes in late anaphase. The midzone collapses into the midbody following ingression of the contractile ring during telophase. The images are of fixed and stained rat kangaroo kidney (PtK) cells, which are presented in lieu of human cells because the small numbers of chromosomes in these cells make the mitotic events clearer. (Previously unpublished images kindly provided by Jennifer Waters, Harvard Medical School.)
9.2.1.3 Gap Phases and Checkpoints Are Points of Decision Making The G1 and G2 gap phases are decision-making periods, during which intracellular and extracellular signals determine whether the cell is prepared to enter the subsequent S-phase and M-phase. During G2-phase, the key signal is intracellular, emanating from the newly replicated DNA. If serious DNA damage has occurred during replication, entry into mitosis is delayed. During G1-phase, many different kinds of extracellular and intracellular information are integrated into the decision to enter S-phase. Extracellularly, the appropriate signal transduction pathways need to be activated by the binding of receptor ligands. The combination of ligands required to proliferate depends on the cell type, and so we generically refer to them in this chapter as “growth factors” (see Chap. 8, Sec. 8.2). In some cell types, G1 arrest can also be imposed by excessive physical contact with neighboring cells. Together, these extracellular requirements define the special niches in which cells typically proliferate in vivo. Intracellularly, serious genomic damage will block cells in G1-phase. Cells lacking nutrients, such as essential amino acids, will also arrest in G1-phase. Supply of nutrients is unlikely to be limiting in normal animal tissues with adequate blood supply but may be limiting within solid tumors (see Chap. 12, Secs. 12.2 and 12.3). In some mammalian cell types, a minimal cell size may also be needed to enter S-phase, a requirement that would help coordinate cell growth with the cell cycle (Jorgensen and Tyers, 2004).
When essential growth factors or nutrients are removed from cells, death by apoptosis or senescence may result (see Sec. 9.4). If the cells do not die, a transition in G1-phase called the restriction point determines the cellular response (see Fig. 9–1). If growth factors or nutrients are withdrawn before the restriction point, cells will enter the quiescent G0 state. If these factors are withdrawn after the restriction point, cells will progress through a full cell cycle before transitioning into the G0 state early in the subsequent G1-phase. The restriction point can occur early or late in G1-phase, depending upon the cell type. In most cancers, control over the restriction point appears to be loosened as the cells proliferate without the appropriate combination of extracellular signals.
9.2.2 Molecular Mechanisms Central to Cell-Cycle Control
The molecular biology of the cell cycle has been explored intensively with important discoveries having been made in yeasts, frog eggs, fruit flies, cultured mammalian cells, and mice. The molecular wiring of the cell cycle has largely been conserved during eukaryotic evolution and employs the full gamut of cellular regulatory mechanisms: transcription, translation, protein degradation, protein localization, protein phosphorylation, and microRNAs. But two molecular mechanisms are particularly central to cell-cycle regulation: cyclin-dependent kinases (CDKs) and E3 ubiquitin ligases.
9.2.2.1 Cyclin-Dependent Kinase Activity Is Tightly Regulated CDKs are the central regulators of the cell cycle. CDK activities largely define the phases of the cell cycle, while changes in CDK activity drive transitions between these phases. CDKs exert these effects by phosphorylating hundreds of different proteins throughout the cell. But the abundance of CDKs is constant throughout the cell cycle. Multiple, overlapping, posttranslational mechanisms ensure that the catalytic activity of different CDKs is highly regulated in space and time (Fig. 9–4A).
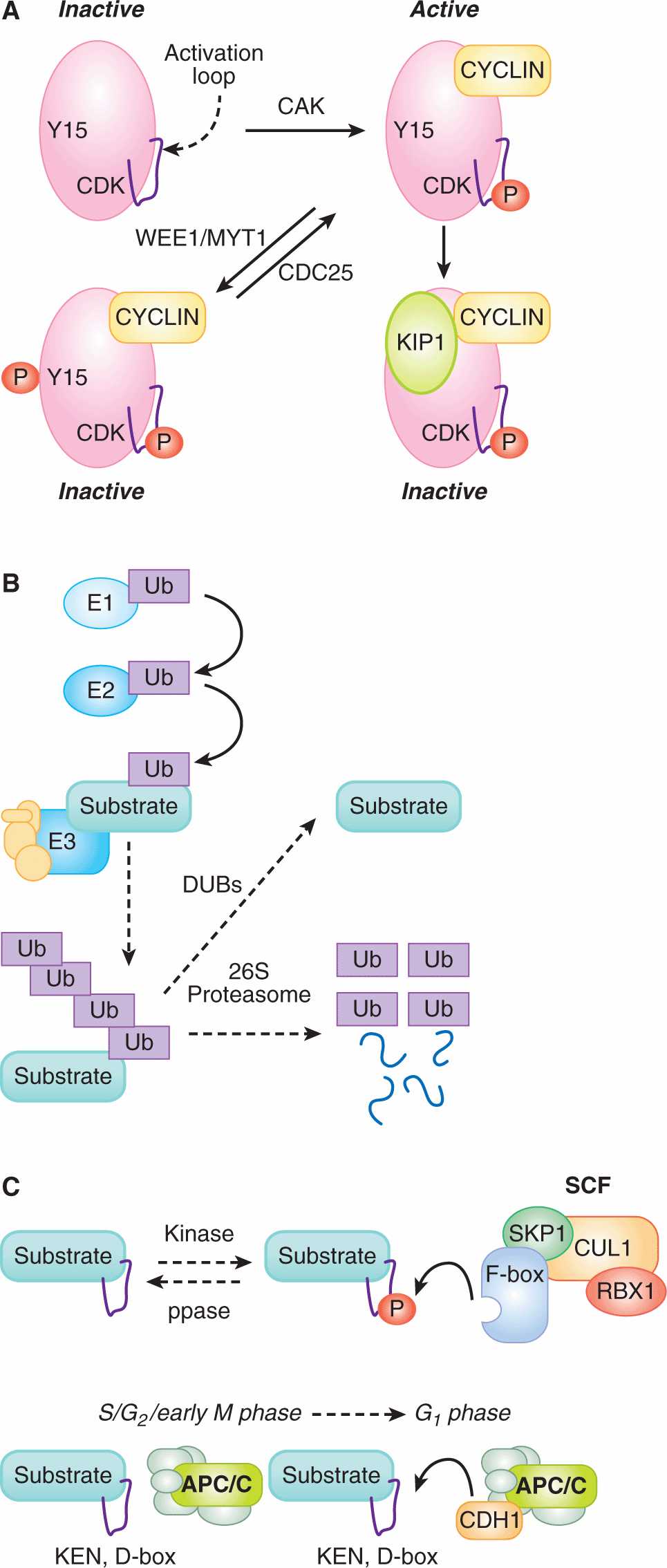
FIGURE 9–4 Molecular mechanisms central to cell cycle control. A) CDKs are the central regulators of the cell cycle. Multiple mechanisms control CDK kinase activity. To become active kinases, CDKs must bind CYCLINs, which leads to the phosphorylation (P) of their activation loop by the CDK activating kinase (CAK). CYCLIN-bound CDKs can, however, become inactivated by phosphorylation on a different residue (Y15) by WEE1/MYT1 kinases. This inhibitory phosphorylation is removed by CDC25 phosphatases. CDKs can also be inactivated upon binding CDK inhibitors (CKIs), such as KIP1. B) Several important cell-cycle proteins are destroyed by ubiquitin-mediated proteolysis. Ubiquitin (Ub) is activated by the E1 ubiquitin-activating protein and the covalent bond then transferred to an E2 ubiquitin-conjugating enzyme. E3 ubiquitin ligases bind to both the E2 and to the substrate protein, catalyzing the formation of a covalent bond between the ubiquitin and the substrate. Iterative rounds of ubiquitination generate a multiubiquitinated substrate, which is recognized by the 26S proteosome and degraded into peptides. If the substrate is also recognized by deubiquitinating enzymes (DUBs), the DUBs may hydrolyze the covalent bond with ubiquitin from the substrate, preempting substrate degradation. C) SCF complexes often recognize substrates when the F-box protein binds to a phosphorylated peptide (degron) in the substrate protein. The rate at which these substrates are degraded is set by the activity of the kinase or the phosphatase (ppase), which is often cell-cycle regulated. In contrast, anaphase promoting complex/cyclosome (APC/C) complexes recognize substrates that contain degrons called D-boxes or KEN-boxes. APC complexes are only active during late mitosis and G1 phase when they have bound to an activating subunit, CDC20 or CDH1.
CDK activity is almost entirely dependent on the binding of a cyclin protein (see Fig. 9–4A). Once bound, a CYCLIN not only greatly stimulates the enzymatic activity of the CDK, it also influences substrate selection by the CDK. Based on sequence similarity, the human genome encodes at least 21 CDKs and 25 CYCLINs, but only a subset of the CYCLIN-CDK pairs have been shown to regulate the cell cycle (Malumbres et al, 2009). In normal cells, CDK1 forms complexes with A- and B-type CYCLINs, CDK2 forms complexes with E- and A-type CYCLINs, and CDK4 and CDK6 form complexes with D-type CYCLINs. In mice and humans, CYCLINs are expressed in small families of 2 to 3 proteins each, with the expression of different isoforms often being tissue-specific. For example, CYCLIN A1 is expressed solely in the male germline in mice, while CYCLIN A2 is expressed in all other cells (Kalaszczynska et al, 2009). CYCLINs D1, D2, and D3 are expressed in different tissues in mice, but with some overlap. CYCLINs also specify the intracellular location of CDK activity, as localized CYCLINs can focus CDK activity to the nucleus, the cytoplasm, or the Golgi apparatus. The location of some CYCLIN-CDK complexes changes dynamically during the cell cycle.
Typically, A-, B-, and E-type CYCLINs oscillate in abundance during the cell cycle, defining windows of potential CDK1 and CDK2 activity. In contrast, the abundance of D-type CYCLINs does not change appreciably during the cell cycle (see Fig. 9–5). When a CYCLIN binds a CDK, a threonine in the activation loop of the CDK is phosphorylated by the CDK-activating kinase (CAK) (see Fig. 9–4A; Merrick et al, 2008). In human cells, CAK is actually another CYCLIN-CDK complex composed of CYCLIN H-CDK7, which is constitutively active throughout the cell cycle (Harper and Elledge, 1998).
CDK activity is further shaped by repressive influences (see Fig. 9–4A). The MYT1 and WEE1 kinases inhibit CYCLIN B-CDK1 complexes by phosphorylating amino acids of CDK1. CDC25 phosphatases remove the phosphates from these amino acids. CDK activity can also be curbed by 2 families of CDK inhibitor proteins (CKIs). The INK4 family (p16INK4A, p15INK4B, p18INK4C, p19INK4D) of CKIs bind to CDK4 and CDK6 and prevent their binding to D-type CYCLINs. The CIP/KIP family (p21CIP1, p27KIP1, p57KIP2) of CKIs are more generalized inhibitors, binding and inhibiting the activity of CYCLIN E-CDK2, CYCLIN A-CDK2, and CYCLIN B-CDK1.
9.2.2.2 Ubiquitin-Mediated Proteolysis and E3 Ligases Degrade Cell-Cycle Regulators Critical cell-cycle events are triggered by the destruction of regulatory proteins. Protein degradation is rapid and irreversible—these properties are advantageous when regulating a dynamic and unidirectional process such as the cell cycle. The primary cellular mechanism for targeted protein degradation is ubiquitin-mediated proteolysis in which the small protein ubiquitin is covalently attached to target proteins (see Chap. 8, Sec. 8.2.8). In this process, an E1 ubiquiting-activating enzyme forms a covalent bond with free ubiquitin and then transfers this bonded ubiquitin to an E2 ubiquitin-conjugating enzyme (see Fig. 9–4B). The ubiquitin is then transferred from the E2 enzyme to the substrate protein, a process that requires the binding of both the E2 enzyme and the substrate protein to the E3 ubiquitin-ligase. Substrate selectivity in ubiquitin-mediated proteolysis appears to be entirely conferred by the binding interaction between the substrate and the E3 ligase. The same substrate can be ubiquitinated multiple times—it is the presence of multiple ubiquitins attached to a substrate protein that result in that protein binding to and being proteolyzed by the 26S proteosome. Deubiquitinating enzymes (DUBs) can, however, hydrolyze the covalent bond between a substrate and ubiquitin, counteracting the actions of the E3 ligase. For any given ubiquitinated protein, the rate of proteolysis by the 26S proteosome is set by the relative activity of the E3 ligase and the DUB (see Fig. 9–4B; Komander et al, 2009).
SCF, CUL4-DDB1, and the anaphase promoting complex/cyclosome (APC/C) are E3 ligases with important cell-cycle roles. These 3 protein complexes are evolutionarily related to one another and share similar modular structures, with a catalytic core binding to multiple substrate binding proteins. The catalytic core of SCF complexes binds, via an adaptor protein (SKP1), to multiple substrate-binding proteins (F-box proteins) (Willems et al, 2004). As individual F-box proteins are typically able to bind many different substrates, SCF complexes catalyze the ubiquitination of a myriad of target proteins. CUL4-DDB1 complexes are similarly organized with a catalytic core which binds, via an adaptor protein (DDB1), to multiple substrate binding proteins (DWD proteins) (Jackson and Xiong, 2009). The structure of the APC/C is more complicated: its catalytic core is bound to at least 11 other proteins, and this complex then binds to substrate-binding proteins (CDC20, CDH1) in a cell-cycle–dependent manner (Hutchins et al, 2010; Peters, 2006).
Despite their structural similarities, SCF and APC/C differ in how they are activated to ubiquitinate their targets during the cell cycle (Peters, 2006; Willems et al, 2004). In both cases, short peptide sequences (degrons) in the targeted proteins are recognized by the E3 ligase complexes. In the case of SCF complexes, F-box proteins frequently recognize degrons only when they include a phosphorylated serine or threonine (see Fig. 9–4C). The SCF complexes are constitutively active, but they cannot target a substrate for degradation until that substrate is phosphorylated. Typically, SCF activity towards a given substrate is dependent on the activity of the kinase that phosphorylates that substrate. In sharp contrast, APC/C E3 ligases are only active during certain phases of the cell cycle. APC/CCDC20 is active during prometaphase and metaphase and APC/CCDH1 is active from anaphase to the end of G1-phase (see Fig. 9–4C). During this time, the APC/C complexes bind to degrons that are common to their substrates, most importantly destruction (D) boxes (both APC/C complexes) and KEN boxes (APC/CCDH1 only) (Pfleger and Kirschner, 2000). Because these degrons are not usually phosphorylated or otherwise modified, the degradation of these substrates doesn’t rely on kinases and simply occurs during the windows of APC activity.
9.2.3 The Molecular Underpinnings of the Human Cell Cycle
The biochemical essence of the cell cycle is an oscillation between 2 states (Nasmyth, 1996). The first state lasts from anaphase to the end of the next G1-phase, while the second state lasts from the beginning of S-phase to metaphase (Fig. 9–5). In the first state, CDK1 and CDK2 activity is low, APC/CCDH1 activity is high, and pre-RC assembly on replication origins is permitted. In the second state, CDK2 activity is high, APC/CCDH1 activity is low, and pre-RC assembly on replication origins is not allowed. Each biochemical state is highly stable as a result of multiple positive feedback loops that continually reinforce that state. Switching between the 2 stable states requires special mechanisms that overcome the positive feedback that maintains each state. The points at which the cell switches between the 2 states, the G1/S transition and the metaphase-anaphase transition, are key points of cell-cycle control. The G2/M transition is a third key point of control at which the high CYCLIN B-CDK1 activity that controls early mitosis first appears.
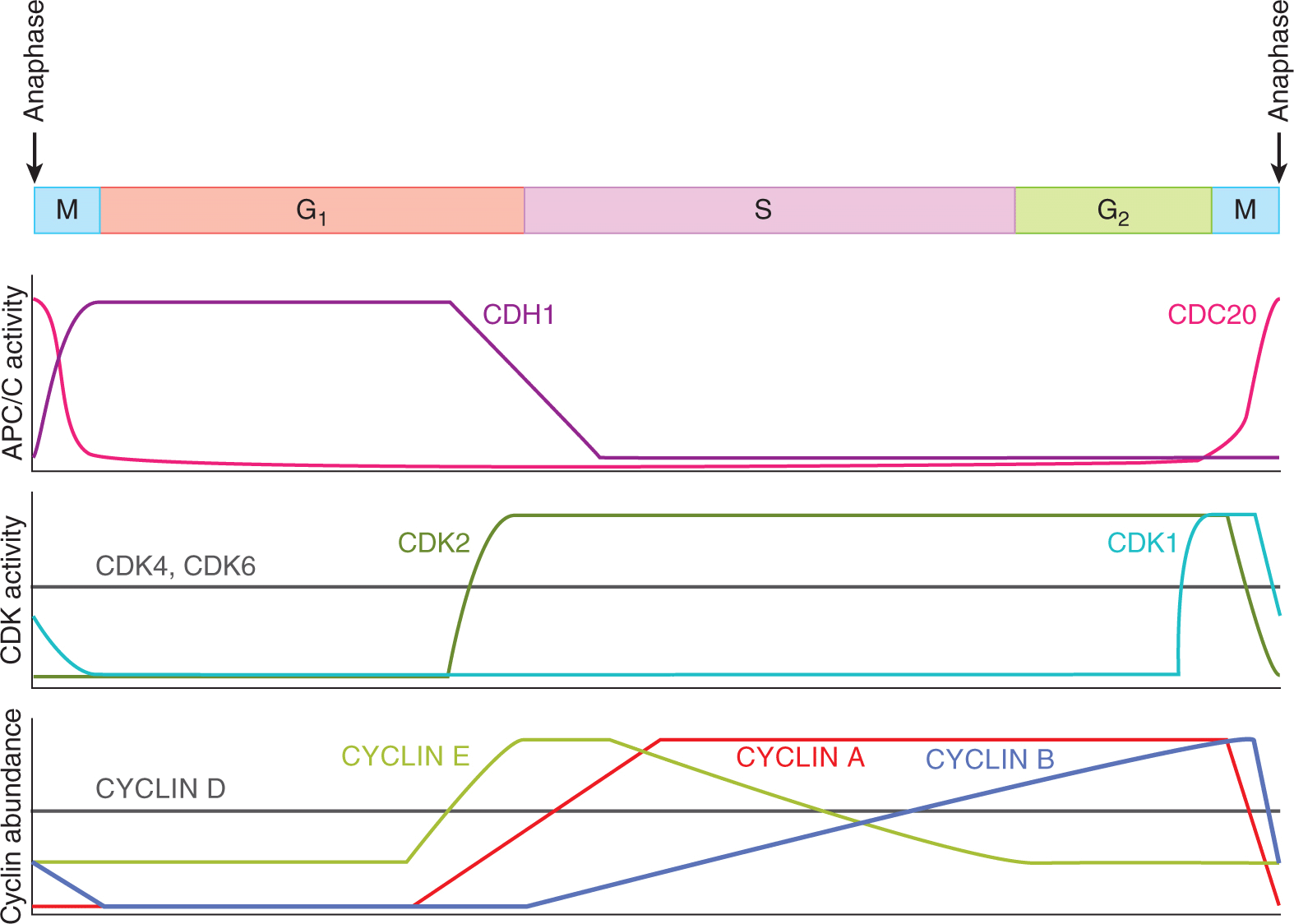
FIGURE 9–5 The pattern of CDC20- or CDH1-dependent APC/C E3 ubiquitin ligase activity, the activity of CDK1, 2, 4, and 6, and the abundance of CYCLINs A, B, D, and E during the cell cycle.
9.2.3.1 G1-Phase Is a Period of Low CDK1 and CDK2 Activity and High APC/CCDH1 Activity Replication origins become capable of initiating DNA replication during G1phase when pre-RCs assemble on them (see Fig. 9–2B; Diffley, 2004). The origin DNA is bound directly by the origin recognition complex (ORC). ORC is then bound by CDC6 and CDT1, which, in turn, recruit MCM complexes to the origin (Arias and Walter, 2007). The absence of CDK1 and CDK2 kinase activity and the high activity of APC/CCDH1 during G1 phase combine to create a biochemical environment that allows pre-RC assembly (see below).
During G1-phase, CDK1 and CDK2 are not bound to CYCLINs and so their kinase activity is very low (see Fig. 9–4A). CYCLIN A2 and CYCLIN B protein levels are very low during G1-phase because both proteins are substrates of APC/CCDH1 and because their genes are transcriptionally repressed (see Fig. 9–5). The fact that CDH1 is itself a substrate of CYCLIN A2-CDK2 and CYCLIN B-CDK1 complexes generates a self-reinforcing positive feedback loop that lasts throughout G1 phase (Fig. 9–6A, inner loop). When phosphorylated by CYCLIN A2-CDK2 or CYCLIN B-CDK1, CDH1 dissociates from the APC/C complex, abolishing APC/C activity. Therefore, the low activity of CDK1 and CDK2 in G1 phase prevents CDH1 from becoming phosphorylated and the activity of the APC/CCDH1 complex remains high, which, in turn, keeps CYCLIN A2 and CYCLIN B levels low. An additional positive feedback loop reinforces low CDK1 and CDK2 kinase activity (Fig. 9–6A, outer loop). In this loop, APC/CCDH1 targets the F-box protein SKP2 for degradation (Wei et al, 2004). As SCFSKP2 targets the CKI p27KIP1 for degradation (Malek et al, 2001; Wei et al, 2004), high APC/CCDH1 activity during G1-phase indirectly stabilizes the CKI p27KIP1. p27KIP1 can then bind to and inactivate any CYCLIN B-CDK1 or CYCLIN A2-CDK2 complexes that do manage to form.
Quiescence or G0 is not simply an indefinitely prolonged G1 period. Like G1-phase cells, G0-phase cells have low CDK1 and CDK2 activity and high APCCDH1 activity. But in contrast to G1-phase cells, G0-phase cells have little to no CYCLIN D-CDK4/6 activity, as a result of sharply repressed CYCLIN D transcription. In G0-phase cells, pre-RCs are not assembled on replication origins, apparently because the abundance of some pre-RC components is very low (Williams et al, 1998). The entry of quiescent cells into G1-phase is a prolonged process that includes the synthesis of D-type cyclins, decreasing levels of p27KIP1, the accumulation of CDC6, CDT1, MCM, and the assembly of pre-RCs. D-type cyclins are transcriptionally induced and p27KIP1 is transcriptionally repressed downstream of growth factor signaling (Sherr and Roberts, 1999; see Chap. 8).
9.2.3.2 The G1/S Transition Marks the Rise of CDK1/2 Activity At the G1/S transition, the positive feedback loops that suppress CDK2 activity and keep the APC/CCDH1 active are overturned (see Fig. 9–6B). CYCLIN D-CDK4/6 complexes are thought to have 2 key roles early in this process. First, CYCLIN D-CDK4/6 functions noncatalytically to bind p27KIP1 protein, and prevents p27KIP1 from inhibiting CYCLIN E-CDK2 and CYCLIN A-CDK2 (Sherr and Roberts, 1999). Second, CYCLIN D-CDK4/6 phosphorylates 3 related “pocket proteins,” the Retinoblastoma (RB) protein, p107, and p130. Phosphorylation of the 3 pocket proteins leads to their release from the E2F family of transcription factors (Fig. 9–7). E2Fs complex with the DP protein and bind to sequences found in the promoters of a broad cohort of genes, many of which encode proteins important for S-phase entry or DNA synthesis. E2F4 and E2F5 bind primarily to p130 and p107, and these complexes act to repress target genes in G0-phase and in early G1-phase. E2F1-3 bind primarily to RB, which keeps E2F1-3 target genes inactive throughout G1-phase (Fig. 9–7). The phosphorylation of the 3 pocket proteins by CYCLIN D-CDK4/6 decreases their affinity for E2Fs, resulting in the inhibitory E2Fs (E2F4, E2F5) leaving the nucleus and the activating E2Fs (E2F1, -2, -3) inducing a broad transcriptional program in late G1-phase (Fig. 9–7).
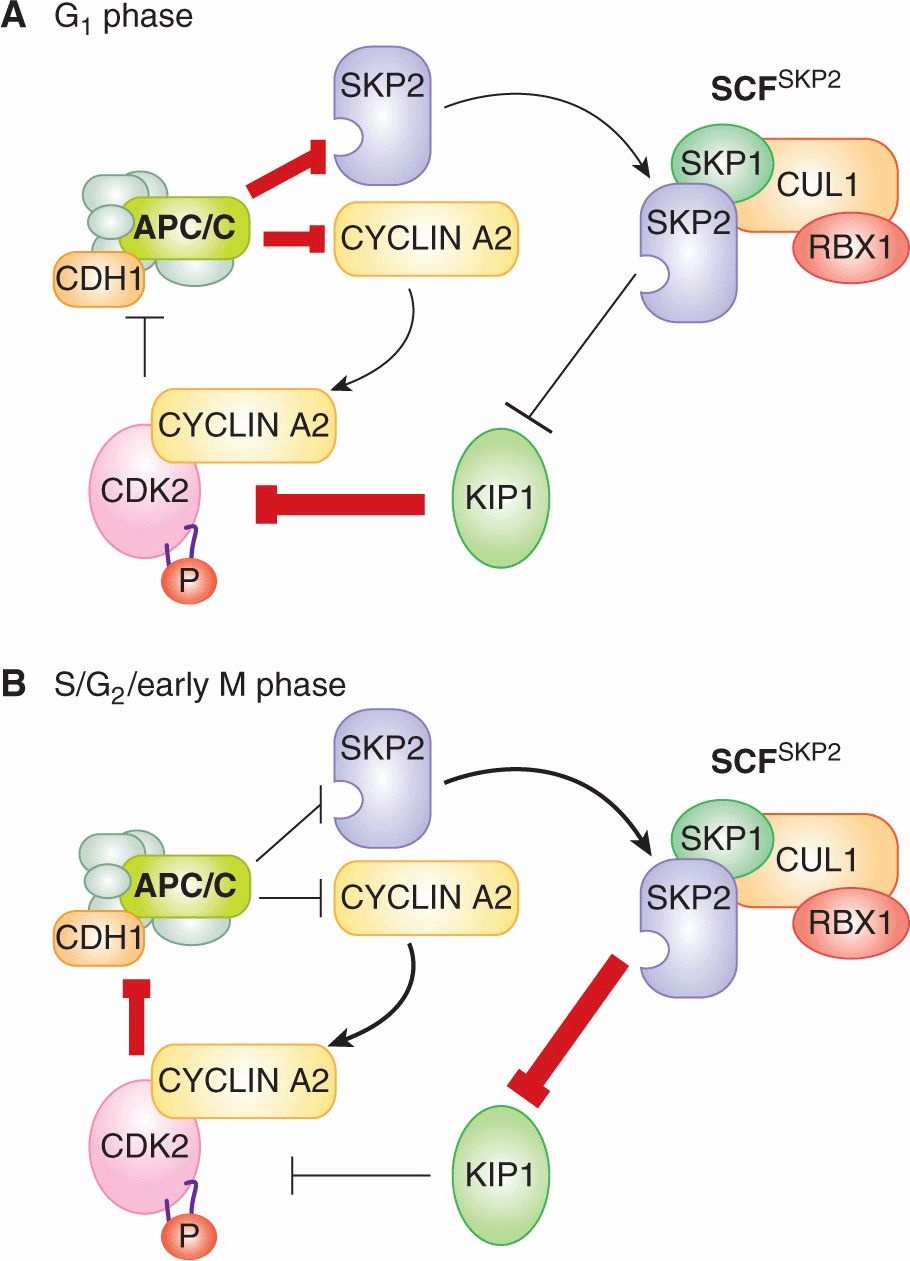
FIGURE 9–6 Positive feedback loops between APC/CCDH1 and CYCLIN-CDK1/2 complexes create 2 self-reinforcing states that help define the 2 primary biochemical states in the eukaryotic cell cycle. A) In G1-phase, APC/C and CDH1 are associated so APC/CCDH1 activity is high. This high APC/CCDH1 activity results in the degradation of CYCLIN A2 and SKP2. Because of the low abundance of these 2 proteins, CYCLIN A2-CDK2 and SCFSKP2 complexes are rare. Low SCFSKP2 activity results in relatively high p27KIP1 abundance. p27KIP1 helps suppress the activity of any CYCLIN A2-CDK2 complexes that do form. CYCLIN B-CDK1 has a similar place to CYCLIN A2-CDK2 in the positive feedback loops, but is not shown for simplicity. B) In S/G2/early M phase, the balance of power is reversed. Now, CYCLIN A2 is abundant, so CYCLIN A2-CDK2 complexes form and suppress APC/CCDH1 activity. Similarly, SKP2 is abundant, so SCFSKP2 complexes form and cause the degradation of p27KIP1
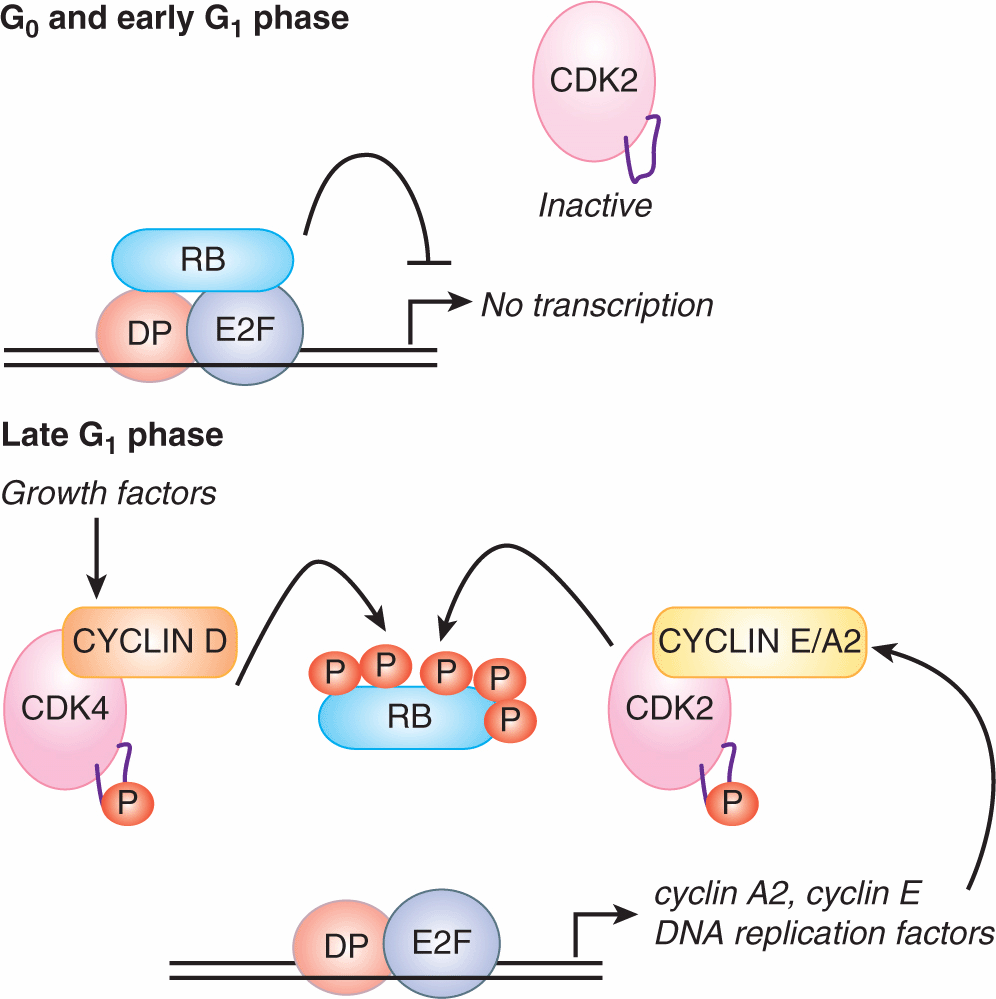
FIGURE 9–7 In late G1 phase, the transcription of cyclin E, cyclin A2, and DNA replication factors is driven by the E2F1-3 transcription factors. Five types of E2F bind to DNA-binding sites in the promoters of hundreds to thousands of genes with the assistance of the DP protein. In G0 and early G1 phases, these genes are repressed by either E2F4-5/p130 complexes (not shown) or E2F1-3/RB complexes. Phosphorylation of the 3 pocket proteins (RB, p130, p107) by CYCLIN D-CDK4/6 causes the pocket proteins to dissociate from the E2Fs during G1-phase, freeing E2F1-3 to activate transcription. The onset of E2F-dependent transcription is also likely to be the molecular basis for the restriction point.
The E2F transcriptional program includes the genes encoding CYCLIN E, CYCLIN A2, and EMI1. Once translated, these 3 proteins collectively overturn the G1 state of inactive CDK1 and CDK2 and active APC/CCDH1. CYCLIN E-CDK2 and CYCLIN A2-CDK2 complexes phosphorylate RB and further stimulate the expression of E2F1-3 target genes in a positive feedback loop (Fig. 9–7). CYCLIN E-CDK2 and CYCLIN A2-CDK2 also phosphorylate CDH1, causing CDH1 to dissociate from the APC/C (see Fig. 9–6A, inner loop). The assault on APC/CCDH1 activity is furthered by EMI1, a binding partner and direct inhibitor of APC/CCDH1 (Di Fiore and Pines, 2008). The loss of APC/CCDH1 activity leads to the accumulation of its substrate SKP2. Rising SCFSKP2 activity in late G1 phase targets p27KIP1 for degradation, contributing to the rise in CYCLIN E-CDK2 and CYCLIN A2-CDK2 activity (see Fig. 9–6A, outer loop). These interlocking feedback loops, which are elaborated late in G1-phase and cause the G1/S transition, stabilize the new S/G2/early M-phase state of high CDK2 activity and low APC/C activity (see Fig. 9–5).
The switch from low to high CDK2 activity and from high to low APC/CCDH1 activity is thought to be the molecular basis of the restriction point. That is, at some point in G1-phase, this switching process becomes irreversible and no longer requires growth signaling pathways. As discussed above, these signaling pathways are thought to drive the G1/S transition by (a) stimulating the synthesis and activity of CYCLIN D-CDK4/6 complexes, and (b) transcriptionally repressing and directly inactivating p27KIP1.
9.2.3.3 The Initiation of DNA Replication and the Block to Overreplication The rise of CYCLIN A2-CDK2 activity at the G1/S transition triggers DNA replication. In at least some cell types, CYCLIN E-CDK2 also contributes to this process (Kalaszczynska et al, 2009). DNA replication also requires the CDC7 kinase, whose activity requires binding to the DBF4/ASK protein. Like CYCLIN A2, DBF4/ASK increases in abundance at the G1/S transition (Jiang et al, 1999). Phosphorylation by CYCLIN A2-CDK2 and CDC7-DBF4 of the pre-RC complex assembled around a particular DNA replication origin leads to the recruitment of replication proteins to that origin. The DNA double helix at the origin is subsequently unwound by the MCM helicase (Takeda and Dutta, 2005). These events attract DNA polymerase complexes, which bind to the unwound ssDNA at the origin and initiate DNA replication (see Fig. 9–2).
The reloading of pre-RC complexes on replication origins in S-, G2-, and early M-phase is prevented primarily by eliminating CDT1 activity (Fig. 9–8A; Arias and Walter, 2007). Overlapping mechanisms are likely necessary to ensure that pre-RC complexes do not reappear on any of the tens of thousands of replication origins over the many hours it takes to complete S, G2, and early M phases. Two different E3 ligases target CDT1 for degradation. In S, G2, and early M phases, phosphorylation of CDT1 by CYCLIN A2-CDK2 leads to CDT1 being ubiquitinated by SCFSKP2. In S-phase, CDT1 also interacts with the chromatin-bound proliferating cell nuclear antigen (PCNA), which also leads to CDT1 becoming ubiquitinated. Any CDT1 that escapes ubiquitin-mediated degradation is inactivated by the Geminin protein. Geminin (GEM) binding to CDT1 prevents the recruitment of MCM complexes to DNA replication origins, thereby blocking a crucial step in pre-RC assembly. Geminin is itself an APC/C substrate that starts to accumulate at the G1/S transition when APC/CCDH1 is inactivated. Therefore, CDT1 and GEM are binding partners that have reciprocal patterns of degradation and abundance during the cell cycle (Fig. 9–8B). In addition to the mechanisms that restrict CDT1 activity in S, G2, and early M phases, CDC6 and components of the ORC complex may also be negatively regulated by CYCLIN A2-CDK2 and CYCLIN B-CDK1 phosphorylation during this same period (Arias and Walter, 2007; Diffley, 2004).
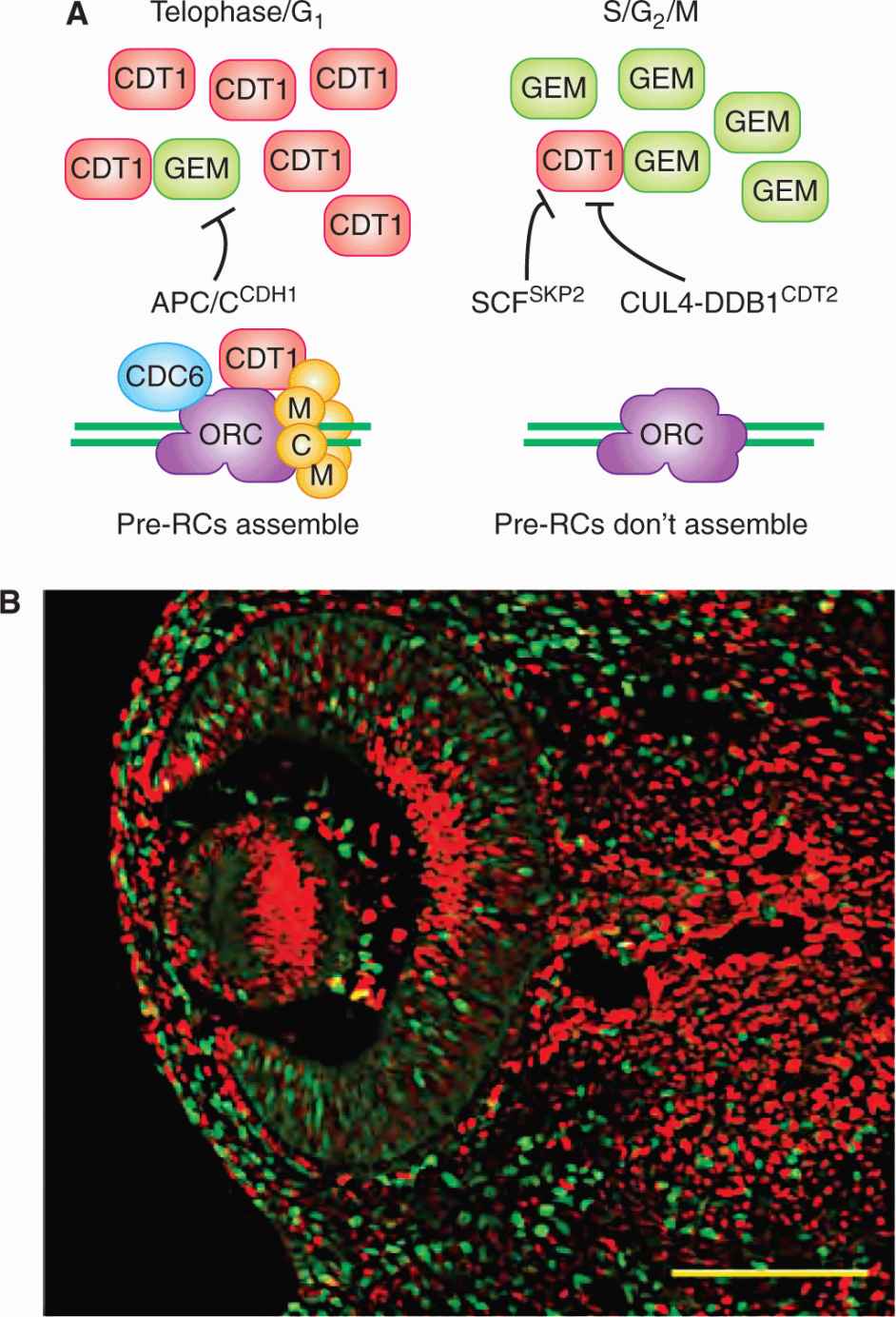
FIGURE 9–8 A) Multiple mechanisms inhibit CDT1 in S-, G2-, and early M-phase, thereby preventing new pre-RCs from assembling during these cell-cycle phases. The protein stability and abundance of CDT1 and Geminin (GEM) are inversely related during the cell cycle. CDT1 is stable and abundant during telophase and G1-phase, while GEM is stable and abundant during S-, G2-, and early M-phase. The E3 ubiquitin ligases that target CDT1 and GEM for degradation in each phase are shown. B) Fluorescent proteins fused to the degron sequences of CDT1 (red) and GEM (green) localize to the nucleus and report the cell-cycle dependent instability conferred by these degrons. In the eye of a mouse that expressed both reporters in all of its cells, it is evident that cells are either red or green, depending on the cell cycle phase of that cell. (From Sakaue-Sawano et al, 2008.)
During DNA replication, the 2 emerging sister chromatids become connected by a protein complex called Cohesin. By keeping the sister chromatids tightly connected, Cohesin is critical for the segregation of the 2 chromatids into separate daughter cells during mitosis. Cohesin is also important for the repair of dsDNA breaks (DSBs) by recombination between sister chromatids (see Chap. 5, Sec. 5.3.4; Peters et al, 2008). Cohesin may link sister chromatids by simply enclosing them within the same ring of protein (Nasmyth and Haering, 2009). How Cohesin establishes sister-chromatid cohesion during S-phase remains uncertain, but it is known to require a cohort of loading and stabilization proteins (Peters et al, 2008).
9.2.3.4 G2 and the Entrance to Mitosis In late G2-phase, mammalian cells experience a surge of CYCLIN B-CDK1 activity that initiates the events of early mitosis. Throughout S- and G2-phase, the concentration of CYCLIN B rises, being induced by the FOXM1 transcription factor (see Fig. 9–5). The newly translated CYCLIN B binds to unphosphorylated CDK1. The binding of CYCLIN B allows phosphor-ylation of CDK1 on the activation loop, generating a momentarily active CYCLIN B-CDK1 kinase (see Fig. 9–4A; Deibler and Kirschner, 2010). This complex is quickly recognized in the cytoplasm by MYT1 and in the nucleus by WEE1, 2 protein kinases that phosphorylate other amino acids (particularly tyrosine 15) of CDK1 and thereby quell the kinase activity of the CYCLIN B-CDK1 complex (O’Farrell, 2001; see Fig. 9–4A). Throughout S- and G2-phase, these inactive, phosphorylated CYCLIN B-CDK1 complexes accumulate in the cell.
In late G2-phase, the stockpiled complexes are suddenly activated by a rapid loss of the inhibitory phosphorylations, a process that is driven by at least 2 positive feedback loops (O’Farrell, 2001; Lindqvist et al, 2009). In the first positive feedback loop, CYCLIN B-CDK1 phosphorylates and activates the CDC25 phosphatases, which remove the inhibitory phosphate groups. In the second positive feedback loop, CYCLIN B-CDK1 inactivates WEE1 and MYT1, thereby blocking any further phosphorylations on CDK1. As inactive CYCLIN B-CDK1 complexes accumulate for many hours during S-phase and G2-phase, some trigger must be required to activate these positive feedback loops in late G2-phase. A strong candidate for the late G2-phase trigger is simply the accumulation of CYCLIN B and/or CYCLIN A2 above a threshold concentration. The time required to achieve this threshold concentration of CYCLIN B and/or CYCLIN A2 would then set the length of G2-phase.
The elaboration of CYCLIN B-CDK1 activity in late G2-phase is blocked by severely damaged or unreplicated DNA, a checkpoint that prevents cells from attempting to segregate damaged chromosomes (see Sec. 9.3 and Chap. 5, Sec. 5.4).
9.2.3.5 The Events of Early Mitosis Culminate in Sister Chromatid Separation The early events of mitosis are orchestrated by 4 mitotic kinases: CYCLIN B-CDK1, Polo-like kinase 1 (PLK1), Aurora (AUR) A, and AUR B. Correspondingly, more than 1000 proteins become phosphorylated specifically during mitosis (Dephoure et al, 2008). All 4 mitotic kinases are activated at the G2/M transition or in prophase. CYCLIN B-CDK1 activation has been discussed. Activation of PLK1, AUR A, and AUR B results from phosphorylation and from binding interactions that localize these kinases to critical locations in the mitotic cell (Fig. 9–9). At the end of mitosis, all 4 of the mitotic kinases are inactivated, in large part as a result of the resurgence of APC/C activity. CYCLIN B is targeted for degradation by APC/CCDC20 during metaphase, while the degradation of PLK1, AUR A, and AUR B by APC/CCDH1 begins in anaphase (Barr et al, 2004; Carmena et al, 2009).
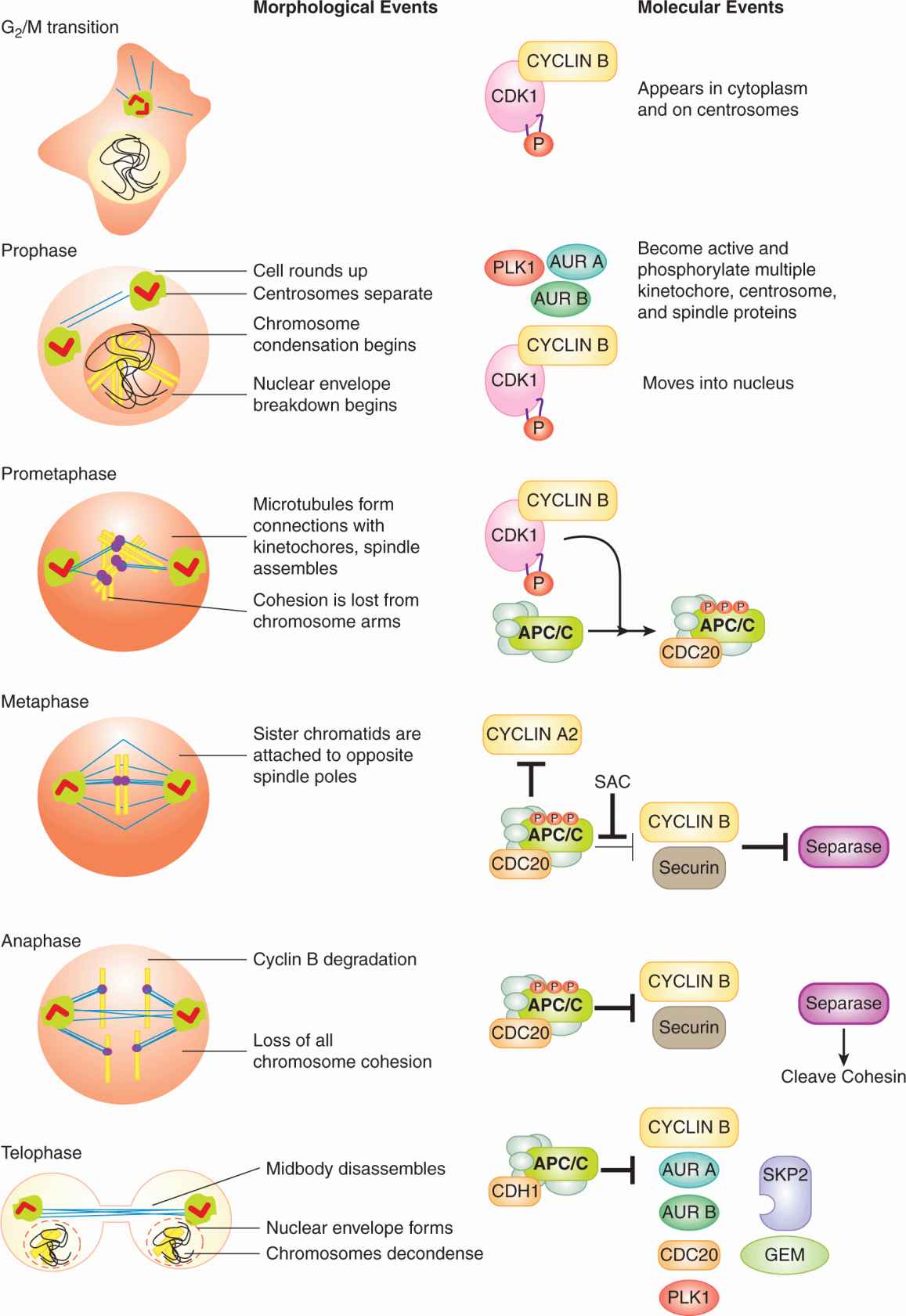
FIGURE 9–9 Major morphological and molecular events of mitosis. Schematic (left) images of cells in the primary stages of mitosis. Uncondensed DNA is in black, condensed DNA is in yellow and microtubules are in blue. Purple spheres on DNA represent kinetochores. Orthogonal red rectangles within green spheres represent centrosomes. The concentration of active CYCLIN B-CDK1 complexes is roughly indicated by the intensity of orange in the cytoplasm and nucleus. See text for details on the major molecular events. P, Phosphorylation; SAC, spindle assembly checkpoint.
The strength and location of CYCLIN B-CDK1 activity during early mitosis helps determine the sequence of early events (see Fig. 9–9; Gavet and Pines, 2010). CYCLIN B-CDK1 activity is first apparent in the cytoplasm and on the centrosomes. In early prophase, cytoplasmic CYCLIN B-CDK1 causes adherent cells to “round up” and, in collaboration with PLK1, causes the 2 centrosomes to separate and mature (Gavet and Pines, 2010). CYCLIN B-CDK1 then moves into the nucleus and by late prophase the complex is predominantly nuclear (Takizawa and Morgan, 2000). Nuclear CYCLIN B-CDK1 triggers chromosome condensation, as well as the breakdown of the nuclear envelope, in part by phosphorylating and disassembling the meshwork of intermediate filaments called lamins that lines the inner nuclear membrane.
Upon dissolution of the nuclear envelope, which marks the start of prometaphase, microtubules gain access to the chromatin. Microtubule interaction with the condensing chromosomes leads to the self-organization of the mitotic spindle between the 2 centrosomes (see Fig. 9–9). This process is highly complex and requires numerous proteins that move and stabilize microtubules (Gatlin and Bloom, 2010). During prometaphase, 20 to 25 microtubules bind the kinetochore of each sister chromatid (Walczak et al, 2010). By metaphase, each sister chromatid pair has formed a bipolar attachment to the mitotic spindle: one sister chromatid’s kinetochore is connected by microtubules to one spindle pole, while the other sister chromatid’s kinetochore is connected by microtubules to the other spindle pole (see Fig. 9–9). Each microtubule bundle exerts pulling forces on the kinetochore toward the spindle pole from which the bundle emanates. Although their kineto-chores and centromeres are being pulled in opposite directions, the sister chromatids do not separate as a result of the Cohesin complexes that continue to physically connect them. The balance of these pulling and resistive forces, amongst other forces present in the mitotic spindle, cause each sister chromatid pair to align at the metaphase plate (Dumont and Mitchison, 2009). By phosphorylating multiple centrosome, spindle, and kinetochore proteins, the 4 mitotic kinases play key roles in shaping the mitotic spindle and aligning sister chromatid pairs (Carmena et al, 2009; Petronczki et al, 2008).
The loss of cohesion between the sister chromatid pairs occurs in 2 steps. During prophase and prometaphase, most of the Cohesin along the chromosomal arms dissociates, allowing the chromosomal arms to partially separate by metaphase (Peters et al, 2008). But, Cohesin located at the centromeres does not dissociate until the onset of anaphase. The 2-step loss of cohesion explains the X-shaped structure of chromosomes in karyotypes, which are derived from cells arrested in metaphase by spindle poisons.
The APC/C becomes active early in mitosis when CDC20 binds to the APC/C following extensive phosphorylation of the APC/C by CYCLIN B-CDK1. The bound APC/CCDC20 is active versus some substrates in prometaphase, including CYCLIN A2 (Geley et al, 2001), but remains inactive toward other critical substrates in prometaphase due to the inhibitory effects of the spindle assembly checkpoint (SAC; see Fig. 9–9). Once all of the kinetochores have attached to microtubules and each sister chromatid pair in under tension, cells are in metaphase, the SAC is inactivated, and the APC/CCDC20 becomes fully active. This fully active APC/CCDC20 then ubiquitinates 3 crucial substrates: securin and the 2 B-type Cyclins (B1 and B2). The proteolysis of these proteins is probably sufficient to explain the loss of sister chromatid cohesion that triggers anaphase (Oliveira et al, 2010). Securin is a binding partner and inhibitor of the protease Separase (Zou et al, 1999). In parallel to Securin, CYCLIN B-CDK1 phosphorylation of separase represses separase activity (Stemmann et al, 2001). Anaphase is initiated when the APC/CCDC20 simultaneously removes the 2 blocks (Securin and Cyclin Bs) to separase protease activity (Oliveira et al, 2010). Separase then cleaves the Cohesin subunit SCC1, which causes the removal of centromeric Cohesin and any Cohesin remaining on the chromosome arms. Once Cohesin is entirely removed, the sister chromatids are free to respond to the pulling forces exerted on their kinetochores and progress toward opposite spindle poles. Note that this current model of anaphase may not explain the synchrony with which the 46 pairs of sister chromatids separate.
9.2.3.6 Late Mitosis Ends with Two Genetically Identical Daughter Cells The events of late anaphase and telophase are largely driven by the loss of protein phosphorylations that were applied earlier in mitosis by the CYCLIN B-CDK1, PLK1, and AUR B mitotic kinases (Sullivan and Morgan, 2007). Also key to late mitosis is the appearance of APC/CCDH1 activity (see Fig. 9–9). With the degradation of CYCLIN B during metaphase and anaphase, CYCLIN B-CDK1 kinase activity is much lower by telophase, allowing CDH1 to become dephosphorylated and bind to the APC/C (see Fig. 9–9). APC/CCDH1 has a much broader substrate range than APC/CCDC20. As 1 of the substrates of APC/CCDH1 is CDC20, APC/CCDC20 activity is lost in late mitosis (see Fig. 9–5).
Many of the molecular details of late anaphase and telophase remain mysterious. The loss of protein phosphates applied earlier in mitosis by CYCLIN B-CDK1—most of which remain unidentified—is required to disassemble the mitotic spindle, decondense the separated chromosomes, and form nuclear envelopes around the 2 daughter genomes. One relatively well-understood event of telophase is the assembly of pre-RCs on DNA replication origins (Arias and Walter, 2007). Here the APC/C has important roles. The ubiquitin-dependent proteolysis of CYCLIN A2 in prometaphase and CYCLIN B1 and B2 in metaphase by APC/CCDC20 reestablishes the low-CDK1/2 activity state required for pre-RC formation (see above). In late anaphase and telophase, APC/CCDH1 maintains the degradation of these CYCLINs. APC/CCDH1 also initiates the destruction of GEM (the binding inhibitor of the pre-RC component CDT1) and SKP2 (which targets CDT1 for degradation via the SCFSKP2 complex) (Wei et al, 2004; Arias and Walter, 2007). As a result of APC/CCDH1 activity, CDT1 can accumulate in telophase, bind to ORC at DNA replication origins, and then recruit the remaining components of the pre-RC. The DNA replication origins are now prepared to initiate DNA synthesis in the next S-phase.
Separation of the daughter cells (cytokinesis) is caused by the gradual constriction of a contractile ring attached to the inner face of the plasma membrane. The contractile force appears to be provided by filaments of Myosin II motor protein interacting with filaments of Actin, although exactly how these filaments are organized to constrict the ring remains uncertain. During anaphase, the contractile ring forms on the inner plasma membrane around the center of the spindle, equidistant from each mass of chromosomes. Typically, the spindle is located in the center of the cell and so the cell divides in half, but asymmetric spindle positioning leading to asymmetric cell cleavage is common during development and in stem cell divisions. During late anaphase and telophase, the spindle’s morphology changes, passing through an intermediate stage called the midzone before being compacted into a bundle of microtubules called the midbody. Ingression of the contractile ring begins in late anaphase and continues throughout telophase, with membrane vesicles fusing with the cleavage furrow to provide the increased plasma membrane required by 2 smaller cells. The midbody spans the 1 to 1.5μm diameter cytoplasmic bridge connecting the 2 future daughter cells after maximal ingression of the contractile ring (Eggert et al, 2006). Membrane vesicles are then directed by the midbody to fill the remaining hole, a process which completes the physical separation of the 2 daughter cells.
9.2.3.7 Caveats to the Current Model of the Mammalian Cell Cycle Studies of mice carrying null alleles of core cell-cycle regulators have raised important caveats about the current model of the human cell cycle described herein. For example, mice lacking both Cdk4 and Cdk6 die late in development as a result of poor proliferation of hematopoietic precursor cells, but most other cell types in the embryo proliferate normally (Malumbres et al, 2009). Second, genes encoding regulatory proteins that are central to this consensus model are not always essential. SKP2, CDH1, p27KIP1, CDK2, CDK4, CDK6, CYCLIN Ds, CYCLIN Es, and CYCLIN As are not required for the proliferation of many cell types in mice (eg, Geng et al, 2003; Kalaszczynska et al, 2009). Even a triple loss of CDK2, CDK4, and CDK6 still allows most cell types in the mouse embryo to proliferate (Santamaria et al, 2007). The surprising mildness of CYCLIN and CDK knockout phenotypes may be because of unusual CYCLIN-CDK complexes (eg, CYCLIN E-CDK1) that form when the usual binding partners are missing (Santamaria et al, 2007). But these mouse knockout studies also hint that current models may be far from complete.
9.2.4 Cell Growth
Although the term growth is often used loosely to refer to proliferation, the term cell growth specifically refers to increases in cell size. Fundamentally, cell growth is a net increase in biomass, primarily proteins, RNA, and membrane lipids. For cells to proliferate and maintain their size, cell growth must, on average, double the size of the mother cell by the time of mitosis. So, in addition to doubling the amount of DNA and the number of centosomes with each cell cycle, a cell must grow sufficiently to double all its other constituent parts—ribosomes, mitochondria, lysosomes, etc.
It remains unclear how intertwined cell growth and the cell cycle are in human cells. Growth of some cells can become uncoupled from the cell cycle, arguing that they are distinct processes. For example, during the formation of the oocytes, which are giant single cells, cell growth occurs in the absence of any cell division. The uncoupling of cell growth and the cell cycle can also occur in somatic cells when the cell cycle is arrested by a DNA damage checkpoint (see Sec. 9.3). Such cell-cycle arrest usually allows cell growth to continue, resulting in oversized cells. In general, cell growth is more limiting for cell proliferation than is the cell cycle, because in animal cells the events of the cell cycle can be accomplished in far less time than it takes to double the mass of the cell. Because different amounts of time are necessary to double cell mass and to complete the cell cycle, coordination between cell growth and the cell cycle must exist at some level or else cell size would fluctuate wildly, which is not observed (Jorgensen and Tyers, 2004).
For a given human cell, cell growth, like the cell cycle, requires the appropriate combination of growth factors to bind to the cell (see Chap. 8, Sec. 8.2). Some of the molecular regulators of cell growth that lie downstream of growth factor signaling are known. c-MYC is a transcription factor encoded by the oncogene most frequently amplified in cancers. c-MYC binds to gene promoters with its binding partner MAX (see Chap. 7, Sec. 7.5.2). The widespread transcriptional program activated by c-MYC overexpression can stimulate cell growth, in part by inducing genes involved in ribosome synthesis, a process that is intrinsically related to cell growth (Eilers and Eisenman, 2008). c-MYC/MAX can also stimulate the cell cycle by activating the transcription of cyclin D1, cyclin D2 and cdk4, while a c-MYC/MAX/MIZ1 complex represses the genes encoding the p21CIP1 and p15INK4B proteins.
The mammalian target of rapamycin (mTOR) and phophatidylinositol-3 kinase (PI3K) signaling network (see Chap. 7, Sec. 7.5.4 and Chap. 8, Sec. 8.2.5) is a central regulator of animal cell growth and proliferation. This network integrates large amounts of extracellular and intracellular information into the decision to activate the AKT kinase and the mTORC1 kinase complex (Guertin and Sabatini, 2007). These 2 kinases then activate multiple downstream processes that drive cell growth, the cell cycle, and block cell death (Guertin and Sabatini, 2007). Genetic alterations that increase AKT and mTORC1 activity can be oncogenic. Mutations in the gene encoding PTEN (see Chap. 7, Sec. 7.6.2), a phosphatase that counteracts PI3K, can constitutively activate AKT and are common in many cancers (eg, ~40% of endometrial cancers, 30% to 40% of glioblastoma multiforme) (Liu et al, 2009; Sansal and Sellers, 2004). Amplification of the genes encoding growth factor receptors upstream of AKT and mTORC1 (eg, ERBB2 see Chap. 7, Sec. 7.5.3) are also common in cancer (Liu et al, 2009). Two rapamycin analogs (temsirolimus and everolimus, small molecular inhibitor of mTORC1) have been shown to be efficacious in the treatment of metastatic renal cell carcinoma and are in clinical trials for other types of cancer.
To grow, cells need to either import or synthesize large amounts of amino acids, nucleotides and fatty acids. In addition, large amounts of adenosine triphosphate (ATP) are required to polymerize these building blocks into proteins, RNA, and membrane lipids. Consequently, growing cells have special metabolic properties. The anabolic state is programmed by growth factor signaling pathways and transcription factors, particularly the PI3K/mTOR network and c-MYC discussed above (Jones and Thompson, 2009). The anabolic state is characterized by a number of features, particularly rapid uptake and metabolism of glucose. The rapid uptake of glucose by some cancer cells reflects their higher rate of glycolysis. Indeed, some cancer cells produce more pyruvate—the end product of glycolysis—than can be oxidized by the tricarboxylic acid (TCA) cycle in the mitochondria. The excess pyruvate is converted to lactic acid and secreted, a phenomenon commonly known as the Warburg effect (see Chap. 12, Sec. 12.3.1). Although usually thought of as an energy generating pathway, glycolysis–via the interlinked pentose phosphate pathway–also has important roles in supplying cellular building blocks, including the reducing equivalent nicotinamide adenine dinucleotide phosphate (NADPH) which is used in many anabolic reactions, the nucleotide precursor ribose-5-phosphate, the phospholipid precursor glycerol-3-phosphate, and the precursors for 4 amino acids (Jones and Thompson, 2009). The need to produce building blocks like NADPH to allow for rapid cell growth may be the molecular explanation for why some cancer cells engage in such rapid glycolysis that lactic acid must be secreted from the cell.
9.2.5 Modifications to Control of Cell Proliferation in Cancer
The first tumor suppressorgene discovered in humans was Rb1, whose loss was found to be the cause of familial retinoblastoma (see Chap. 7, Sec. 7.6.4). Soon after, cyclin D1 was isolated as an oncogene in a subset of parathyroid adenomas. The cdkn2a locus, which contains the genes encoding the p15INK4B protein, the p16INK4A protein, and the p53 activator ARF, was then identified as a tumor suppressor in familial melanomas. These early findings anticipated a general truth about human cancers, which is that the vast majority contain genetic or epigenetic alterations that loosen control over the G1/S transition. Typical alterations include the amplification of genes that encode proteins that drive the G1/S transition (eg, cyclin D1), as well as genomic deletions, point mutations, and promoter methylations that remove G1/S inhibitors like RB and p16INK4A. In a genome-wide survey of 26 human cancer types, the tightly opposed loci encoding p15INK4B and p16INK4A/ARF were found to be the most frequently deleted genomic region, while the genes encoding CYCLIN D1 and CDK4 were among the 4 most frequently amplified regions (Beroukhim et al, 2010). When genetic alterations to the G1/S machinery are considered collectively, the majority of cancers contain at least 1 such change. For example, more than 90% of liver, ovarian, testicular, and lung cancers bear alterations in the genes encoding the currently known G1/S regulatory apparatus (Malumbres and Barbacid, 2001). Alterations to the genes encoding the G1/S regulatory apparatus are thought to allow cancer cells to enter the cell cycle precociously, defying the extracellular signals that limit the proliferation of their normal neighbors.
A detailed understanding of the cell cycle could be important for developing better anticancer drugs. There is active preclinical development and clinical evaluation of agents designed to inhibit cell-cycle regulators. For example, clinical trials are evaluating specific inhibitors of EG5, PLK1, and the Aurora kinases as such drugs may kill mitotic cells without affecting microtubules in quiescent cells. Similarly, substantial effort has been dedicated to generating CDK inhibitors (Malumbres et al, 2008). Future generations of anticancer drugs could exploit the common alterations to the cell-cycle machinery shared by many cancers, such as defective control over the G1/S transition, loss of the p53 response, aneuploidy, or the presence of extra centrosomes. In particular, cancers may acquire a dependence on nonessential cell-cycle components like D- and E-type CYCLIN-CDK complexes, which could be exploited by targeted therapies. Supporting such an idea, mice lacking cyclin D1 or cdk4 can be highly resistant to tumor formation.
9.3 GENOMIC INSTABILITY AND CHECKPOINT PATHWAYS
Nearly all cancers show evidence of genomic instability. Mutations that increase genomic instability are probably selected early in the evolution of many cancers. Consistent with this idea, many familial cancer syndromes are caused by mutations in genes encoding DNA repair proteins and checkpoint proteins (see Chap. 5, Sec. 5.5 and Chap. 7, Sec. 7.2). Genomic instability can occur through point mutations, the amplification or deletion of genomic regions, and the fusion, loss, or gain of whole chromosomes. The duplication and segregation of the genome during the cell cycle inexorably increases the risk of genomic instability. Checkpoints that arrest the cell cycle in response to DNA damage or to difficulties in segregating the chromosomes exist to alleviate such risks.
9.3.1 The DNA Damage Checkpoints
The cell cycle is intimately associated with the detection and repair of DNA damage. (see Chap. 5) When the cell sustains genomic damage beyond a threshold level, the cell cycle arrests. Arresting the cell cycle gives time to repair this damage before entering either S-phase or mitosis, thereby preventing the replication or segregation of compromised chromosomes. Cells with severely damaged DNA can undergo malignant transformation, as demonstrated by the many hereditary cancer syndromes caused by defective DNA checkpoint and repair genes (see Chap. 5, Sec. 5.5). To protect the body from this threat, a cell’s response to DNA damage can often end in that cell’s death (see Sec. 9.4).
There is a 2-way flow of information between the DNA damage response and the cell cycle. Serious DNA damage arrests the cell cycle, but the cell cycle also regulates the response to DNA damage. Mechanisms that correct common chemical changes to DNA bases, such as base excision repair and nucleotide excision repair, operate constitutively. In contrast, a cell’s response to DSBs, likely the most dangerous type of DNA damage, can depend on its cell-cycle phase. During G1-phase, cells primarily repair DSBs through a nonhomologous end-joining (NHEJ) process, in which broken ends are directly ligated together, often leading to the loss of base pairs. During S- and G2-phases, cells continue to repair DSBs by NHEJ, but also deploy homologous recombination (HR), which typically uses the replicated sister chromatid as a template for correcting damaged DNA. Unlike NHEJ, HR doesn’t usually lead to any genomic instability (see Chap. 5, Sec. 5.3).
Serious DNA damage like DSBs activate DNA damage kinases that trigger cell-cycle arrest through phosphorylation of proteins that control the G1/S and the G2/M transitions. These phosphorylation events lead to the stabilization and activation of both the transcription factor p53 and the CKI p21CIP1. p53 induces a broad transcriptional program of DNA repair proteins (Zilfou and Lowe, 2009). p21CIP1 suppress CYCLIN E-CDK2 and CYCLIN A2-CDK2 activity, preventing entry into S-phase. For cells in S- and G2-phase, DNA damage kinases inactivate all 3 CDC25 phosphatases and activate the WEE1 kinase. These actions arrest cells before the G2/M transition point by preventing the surge of CYCLIN B-CDK1 activity that drives cells into mitosis (Reinhardt and Yaffe, 2009).
9.3.2 The Spindle Assembly Checkpoint
The SAC is essential for the human cell cycle, serving to delay anaphase until all the replicated chromosomes are bi-orientated and under tension (Musacchio and Salmon, 2007). The primary defect detected by the SAC is kineto-chores that are not attached to microtubules. A single unattached kinetochore can prevent anaphase in mammalian cells (Rieder et al, 1995). Properly bi-orientated pairs of sister chromatids will also experience tension between their cohered centromeres, as the 2 microtubule bundles pull the 2 kinetochores in opposite directions. Insufficient levels of tension between centromeres can also lead to activation of the SAC, although the exact mechanism is unresolved (Musacchio and Salmon, 2007).
The SAC blocks anaphase by preventing the full activation of APC/CCDC20. The result is stabilized Securin and CYCLIN B, which can suppress Separase and maintain the metaphase state for a long, though not indefinite, period of time (Brito and Rieder, 2006).
9.3.3 Mitotic Errors, Aneuploidy, and Oncogenesis
In the multihit genetic model of oncogenesis, it is evident that genomic instability that arises during S-phase can contribute to cancer formation. For instance, errors by DNA polymerases can introduce point mutations into genes encoding tumor suppressors, while DSBs can cause chromosomal translocations that lead to the misexpression of oncogenes. But cancers are marked not just by such discrete genetic changes, but also by wholesale changes in the number of chromosomes (aneuploidy), which presumably arise from errors in mitosis. More than 90% of clinically detected solid tumors are aneuploid. Although they typically have less bizarre karyotypes than solid tumors, hematopoietic cancers also frequently show loss or gain of one or more chromosomes (Weaver and Cleveland, 2006).
Despite the strong connection between aneuploidy and solid tumors, it is not clear that aneuploidy is a cause, as opposed to a consequence, of cancer. Aneuploidy is generally detrimental to the proliferation of normal cells, presumably because it creates subtle imbalances in the expression levels of the hundreds or thousands of genes present on a supernumery chromosome. But as a general rule, molecular mechanisms proposed to cause cancer are thought to increase—not decrease—the rate of cell proliferation. How then could aneuploidy be a cause of cancer? First, in contrast to the general presumption that drivers of cancer must increase the rate of cell proliferation, cell proliferation in solid tumors is often considerably slower than in normal renewing tissues (see Chap. 12, Sec. 12.1). Second, populations of aneuploid cells will have greater genetic variability and presumably greater phenotypic variability. In some circumstances, the altered expression of multiple genes resulting from certain combinations of chromosome numbers may generate advantageous phenotypes. Such phenotypic diversity could allow aneuploid populations of cancer cells to rapidly evolve in response to physiological stresses, such as hypoxia or immune responses, or to medical interventions, such as chemotherapy.
When misregulated, several proteins involved in mitosis have been shown to induce tumors in mice. These results suggest that the aneuploidy that results from mitotic errors can be a cause of cancer. Misregulation of mitotic proteins can cause aneuploidy by at least three interconnected mechanisms: chromosomal instability, extra centrosomes, and tetraploidy (Weaver and Cleveland, 2006).
Chromosome instability is the failure to properly segregate 1 copy of the sister chromatid pairs to each of the 2 daughter cells, a failure that results in 2 aneuploid daughter cells. In normal cells, missegregation of a chromosome—leading to 1 daughter cell inheriting both sister chromatids—is a rare event, occurring once every 50 to 100 cell cycles. But in some cancer cell lines, chromosomes can be missegregated nearly every cell cycle (Thompson and Compton, 2008).
Extra centrosomes are a common feature of cancers. In normal cells, centrosomes are similar to chromosomes in that they are replicated during S-phase and then segregated during mitosis. In fact, cells appear to use a similar strategy to ensure that both chromosomes and centrosomes replicate only once per cell cycle: there are distinct windows of time for becoming competent for replication and for actually undergoing replication. The 2 centrioles in a centrosome become competent for replication in telophase when they disengage from each other. Upon entering S-phase, each of the disengaged centrioles initiates construction of a new orthogonal centriole, thereby generating two pairs of centrioles and hence 2 centrosomes. But, once cells have entered S-phase, the pairs of centrioles cannot become competent for another round of replication until they disengage in the subsequent telophase. Therefore, in normal cells, centrosomes are only duplicated once per cell cycle. But, cell-cycle defects that weaken the blocks against centriole rereplication appear to arise during the evolution of many cancers (Nigg, 2002). At the start of mitosis, the presence of extra centrosomes results in a spindle with more than 2 poles being formed. If such a multipolar spindle is stable, the resulting anaphase typically generates 3 or more daughter cells. In such cases, the daughter cells usually die as a consequence of profound aneuploidy. Frequently, however, multipolar spindles collapse into bipolar spindles prior to anaphase, with the extra centrosome clustered around 1 of the 2 spindle poles (Ganem et al, 2009). Such divisions generate only 2 daughter cells but often result in chromosome missegregation as a result of additional attachments being made to kinetochores by the extra centrosome.
Tetraploid cells can arise when anaphase or cytokinesis fail. Some tissues, such as the liver, intentionally give rise to tetraploid cells. But in most cell types, tetraploidy usually results from errors. Because centrosomes are not segregated when tetraploid cells form, tetraploid cells almost always have excessive centrosomes, which may help explain the high rates of chromosomal instability in tetraploid cells (Ganem et al, 2007).
9.4 CELL DEATH
The balance between cell viability and cell death is critical for all organisms, and involves a network of proteins and communication amongst several signaling pathways. Impaired cell death is associated with various human pathologies, including immunodeficiency, autoimmunity, neurodegenerative diseases and cancer. Similar to proliferation, cell death is also essential for embryonic and postnatal development and for tissue homeostasis.
Different types of cell death occur in mammalian cells, including apoptosis (or programmed cell death) and necrosis. Autophagic cell death has been also reported, but current evidence supports a more prominent role of autophagy in cell survival rather than cell death (see Chap. 12, Sec. 12.3.7; Kroemer and Levine, 2008). While initially most of the focus has been placed on apoptosis, which is a highly regulated mechanism involving interplay among a large number of proand antiapoptotic proteins, mounting evidence has emerged to support the genetic regulation of necrosis. Although many proteins have been shown to play critical roles in the different types of mammalian cell death mechanisms, recent studies have unraveled additional functions for a number of these proteins in other important cellular processes.
9.4.1 Morphological Changes Associated with Different Types of Cell Death
Morphological changes within dying cells provide important criteria to distinguish between apoptosis and necrotic cell death. Changes associated with apoptosis include rounding up of the cell, plasma membrane blebbing, cytoplasm shrinkage, alteration of membrane asymmetry, and condensation and fragmentation of the nucleus (Fig. 9–10A). Cells at late stages of apoptosis become fragmented into apoptotic bodies that are eliminated by phagocytic cells without triggering inflammation.
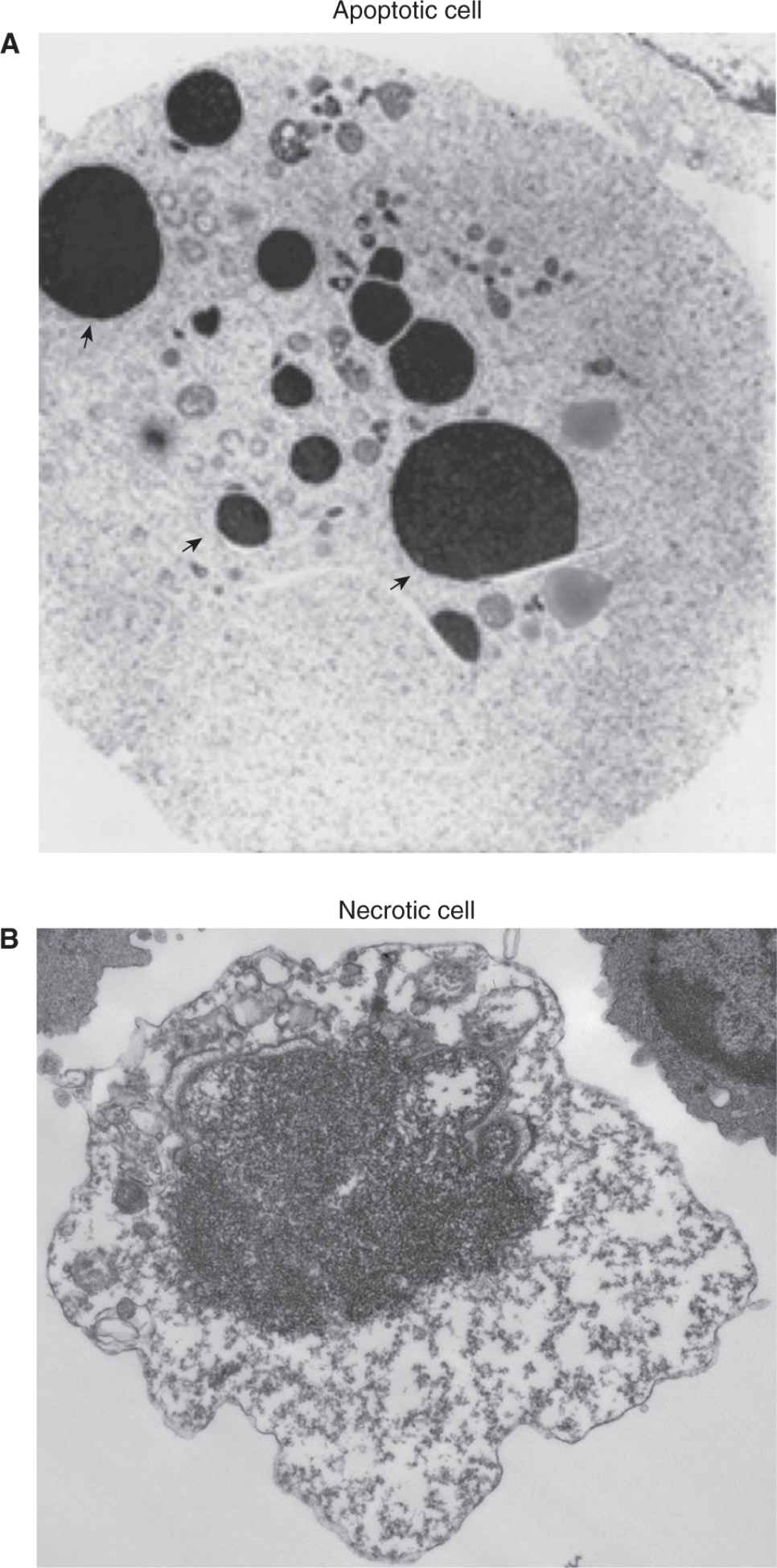
FIGURE 9–10 Morphological changes within dying cells. Transmission electron microscopy images of apoptotic (A) and necrotic (B) cells. Arrows indicate fragmented and condensed nuclear chromatin.
Necrosis-associated cellular changes are manifested by swelling of the cell, mitochondria and cytoplasmic organelles, followed by focal rupture of the plasma membrane. Moderate chromatin condensation is also displayed by necrotic cells. More advanced stages of necrosis are associated with disintegration of all cellular components and inflammation (see Fig. 9–10B).
Transcriptional changes, protein levels and posttranslational modifications can also help differentiate among the different types of cell death.
9.4.2 Mechanisms of Apoptosis
The first evidence that apoptosis is genetically regulated arose from studies of the nematode Caenorhabditis elegans (Kinchen and Hengartner, 2005). During the development of this invertebrate, 131 of the 1090 somatic cells died by apoptosis, and genetic screens identified several genes required for this process. Loss-of-function mutations of C. elegans egl-1, ced-3 or ced-4, or gain-of-function mutations of ced-9 result in survival of the 131 cells programmed to die. The Ced-4 proapoptotic protein binds to Ced-3 and Ced-9, and genetic studies demonstrated that Ced-4 functions downstream of Ced-9 but upstream to Ced-3 (Fig. 9–11). These genetic studies of apoptosis in C. elegans have contributed remarkably to our knowledge of the mechanisms of apoptosis in mammalian cells. In 2002, Sydney Brenner, Robert Horvitz, and John Sulston were awarded the Nobel Prize in Medicine or Physiology for their elucidation of the genetic regulation of organ development and programmed cell death in C. elegans.
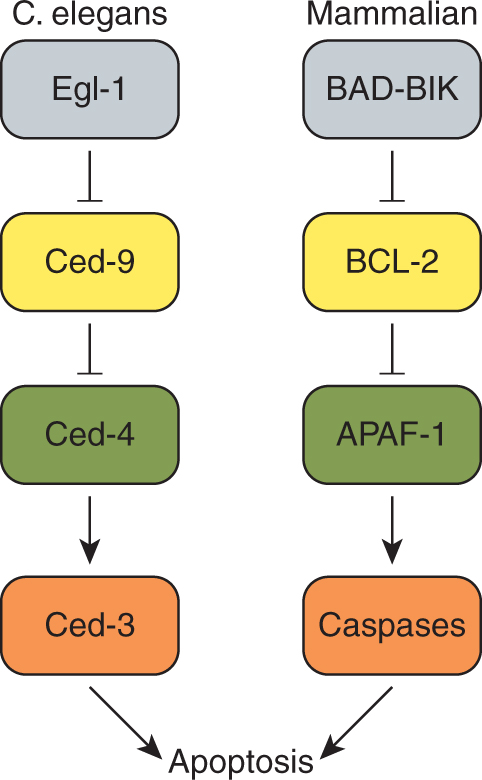
FIGURE 9–11 Major regulators of apoptosis in C. elegans and their mammalian orthologs. Schematic of the major components of the apoptotic pathway in C. elegans (left). Mammalian cells have evolved several orthologs to the pro- and antiapoptotic proteins of C. elegans (right; see text).
Apoptosis is triggered in mammalian cells in response to endogenous stimuli (eg, growth factor deprivation) or exogenous stimuli (eg, irradiation or genotoxic chemotherapeutic drugs). Apoptosis is also induced in response to inadequate cell–matrix interactions and this specific type of apoptosis is known as anoikis. In addition, programmed cell death is also critical for embryonic development, and for the elimination of autoreactive T and B cells (Conradt, 2009).
Biochemical and genetic studies have demonstrated the existence of multiple orthologs of the C. elegans apoptotic proteins Ced-9, Ced-4, and Ced-3. These studies also demonstrated the existence of 2 major apoptotic pathways in mammalian cells; the death receptor (also known as extrinsic) apoptotic pathway and the mitochondrial (also known as intrinsic) apoptotic pathway (Fig. 9–12). Mammalian apoptotic pathways are highly controlled and their regulation involves various proteins including anti-apoptotic (eg, BCL-2 and BCL-XL and proapoptotic (eg, caspases “CASP,” FAS, BIM, and BAX) proteins (Fig. 9–13).
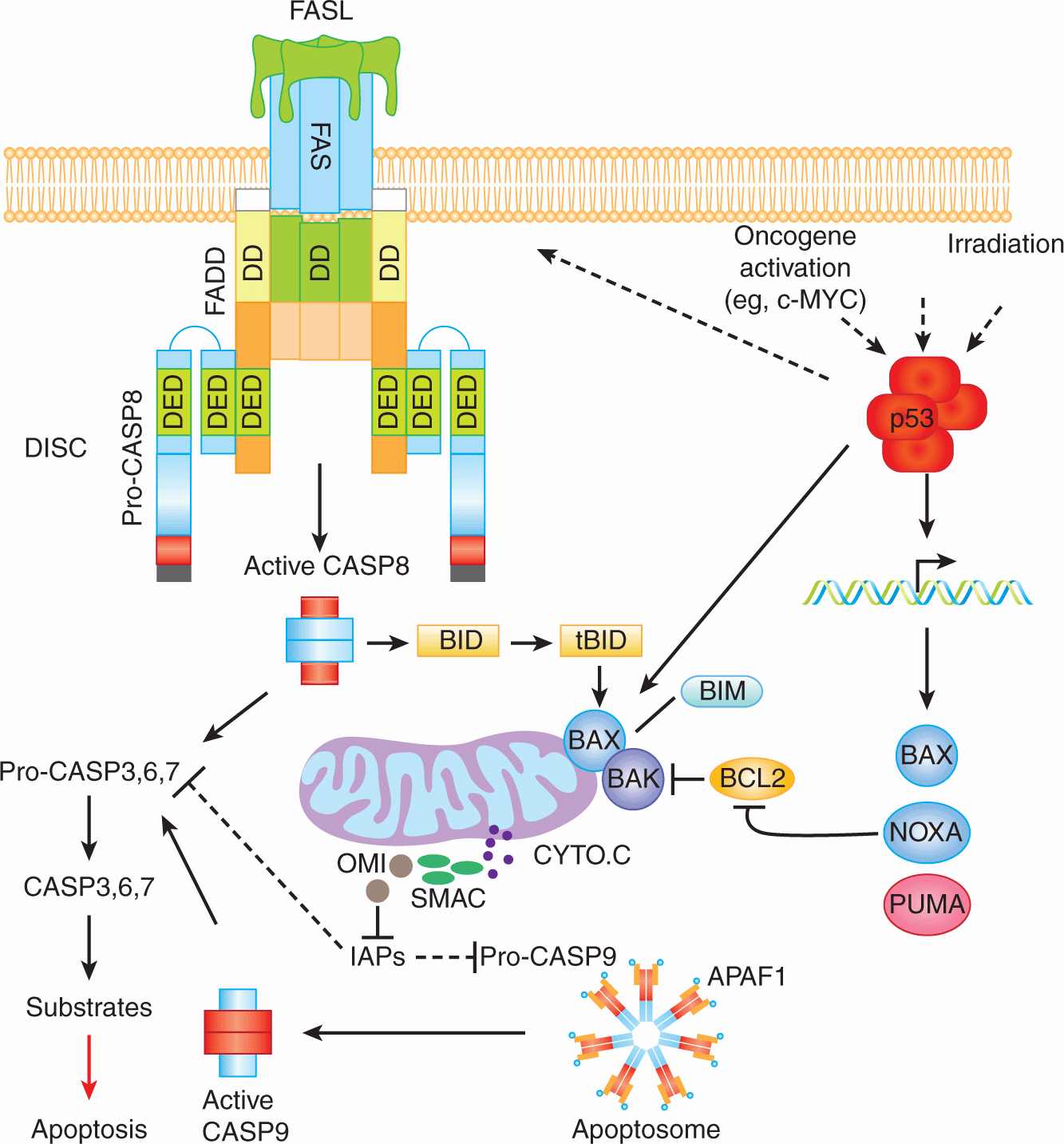
FIGURE 9–12 Schematic of the 2 major apoptotic pathways in mammalian cells. The death receptor pathway is exemplified by the events that occur following engagement of FAS (CD95) by its ligand FASL (CD95L). FAS/FASL interaction leads to the trimerization of the FAS receptor and the recruitment of the adaptor protein FADD to the cytoplasmic tail of FAS. The interaction FAS/FADD is mediated by their respective death domains (DD). This interaction allows the recruitment of CASP8 (and CASP10 in human cells) and the formation of the death-inducing signaling complex (DISC). The presence of these caspases in the DISC results in their oligomerization, autoactivation and subsequent processing of downstream effector caspases (CASP3, -6, and -7). Once activated, effector caspases cleave various cellular proteins leading to apoptotic cell death. The mitochondrial apoptotic pathway is triggered in response to various stimuli including oncogenic activation and DNA damage. Activation of the tumor suppressor and transcriptional factor p53 in response to cellular stresses leads to the transcriptional activation of several proapoptotic genes (eg, BAX, PUMA, NOXA, and FAS). Activation of BAX and BAK leads to outer membrane permeabilization and the release to the cytosol of of a number of mitochondrial proteins including Cytochrome C (CYTO.C). CYTO.C clusters with APAF-1 and the pro-CASP9, forming the apoptosome. The oligomerization of pro-CASP9 within the apoptosome leads to its activation and the subsequent processing of downstream caspases and cell death. In certain cell types, the cleavage of BID by CASP8 generates tBID, a truncated form of BID that translocates to the mitochondria and cooperates with BAX-BAK in inducing the mitochondrial apoptotic pathway.
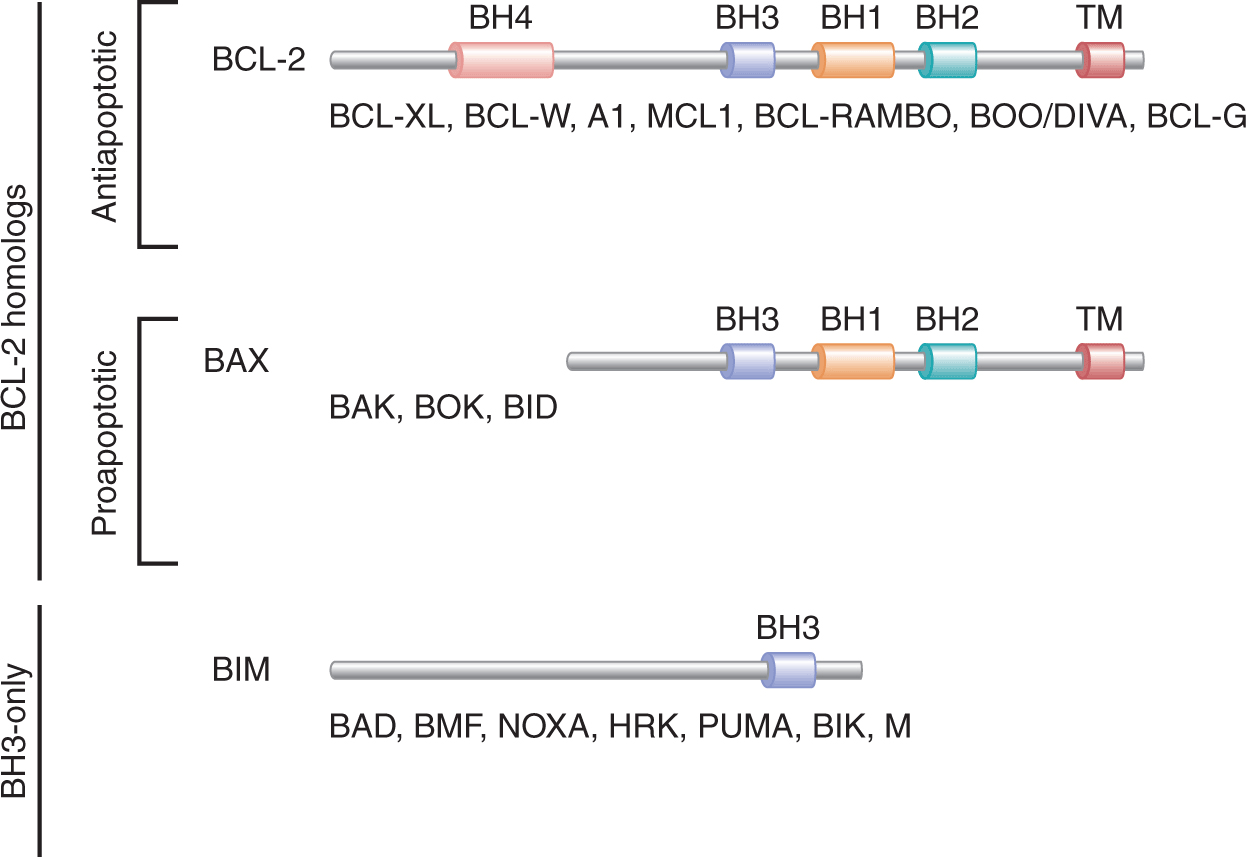
FIGURE 9–13 BCL-2 family members include both anti- and proapoptotic proteins and are classified into different groups. The antiapoptotic group of BCL-2 homologs (eg, BCL-2, BCL-XL, and MCL1) contain 4 BCL-2 homology (BH) domains (BH1 to BH4), and a transmembrane domain (TM). Members of the proapoptotic group of BCL-2 homologs (eg, BAX, BAK, and BID) contain TM, BH1, BH2, and BH3, but lack BH4. Finally, the BH3-only proteins (eg, BIM, NOXA, and PUMA) lack all the domains of the BCL-2 family members with the exception of BH3.
Biochemical studies have proven instrumental for the identification and characterization of the components involved in the initiation, propagation, or inhibition of the mammalian apoptotic signaling pathways. Genetic studies in mice possessing a disruption within, or overexpression of, specific pro- or antiapoptotic genes have been critical to demonstrate the in vivo apoptotic functions of these proteins, and have helped to identify developmental defects and diseases associated with impaired apoptosis.
9.4.2.1 BCL-2 Family Members and Their Roles in Apoptosis The mammalian Bcl-2 gene, a homolog of the C. elegans Ced-9, is a prototypical member of the BCL-2 family that includes antiapoptotic proteins such as BCL-2, BCL-XL, BCL-W, A1, and MCL-1, and proapoptotic proteins, such as BAX, BCL-XS, BAK, BAD, BIK, BIM, BID, and PUMA (see Fig. 9–13; Tait and Green, 2010). Proteins of the BCL-2 family are essential for the initiation and regulation of mammalian apoptosis. These proteins share several conserved domains known as “BCL-2 homology” (BH) regions including BH1, BH2, BH3, and BH4. These domains allow the formation of homo- and heterodimers between BCL-2 family members and are essential for the function of these proteins. BCL-2 family members play important roles in the regulation of apoptosis. For example, overexpression of BCL-2 in mammalian cells inhibits the release of Cytochrome C as well as other factors important for apoptosis (eg, apoptosis-inducing factor [AIF], SMAC, and OMI) from the mitochondrial intermembrane space into the cytosol. Overexpression of BCL-2 also prolongs the survival of cells and increases their resistance to a variety of apoptotic stimuli, including glucocorticoids, phorbol esters, ionomycin, anti-CD3 monoclonal antibodies, irradiation, and chemotherapeutic agents.
9.4.2.2 Role of Caspases in Apoptosis Mammalian caspases, “cysteine-dependent aspartate specific proteases,” are the orthologs of the C. elegans protein Ced-3. The first caspase (ICE or CASP1) was identified in 1993 on the basis of its similarity to Ced-3. More than 14 mammalian caspases have since been cloned (Fig. 9–14; Li and Yuan, 2008). Caspases are present in the cytosol in their inactive forms (procaspases or zymogens). To be activated, they require proteolytic cleavage at specific aspartate residues. Active caspases are heterotetrameric complexes composed of 2 large subunits (~20 kDa) and 2 small subunits (~10 kDa) (see Fig. 9–14). All caspases contain an active site pentapeptide (QACXG; X is R, Q, or G). Although caspases are primarily known for their apoptotic function, caspases such as CASP1 and CASP11 play important roles in inflammation. Mounting evidence supports that CASP8 also possesses nonapoptotic functions (see below).
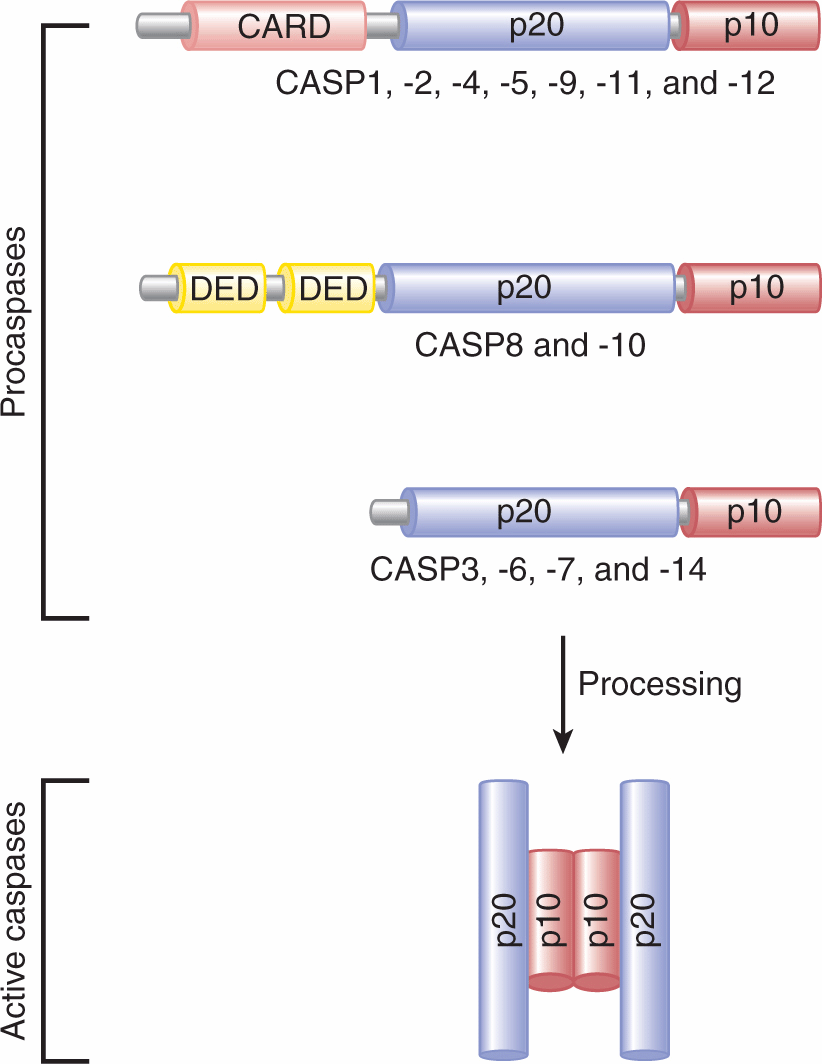
FIGURE 9–14 Mammalian caspases. Caspases exist in an inactive form (procaspases) that require autoprocessing or proteolytic cleavage by upstream caspases in order to be activated. The first group of caspases (CASP1, -2, -4, -5, -9, -11, and -12) contain caspases that contain caspase recruitment domain (CARD) and the subunits p20 and p10. The second group include CASP8 and CASP10, which possess 2 N-terminal death effector domains (DEDs) and the subunits p20 and p10. The third group contains CASP3, -6, -7, and -14. These caspases contain only the 2 subunits, p20 and p10. The processing of procaspases allows the formation of active caspases that consist of heterotetrameric complexes composed of 2 p20 and 2 p10 subunits.
Caspases possess a wide range of expression patterns throughout mammalian tissues. The finding that caspases are able to sequentially process and activate other caspases, together with the structural studies of these proteins, has allowed the classification of caspases into “initiators” (eg, CASP8, CASP9 and CASP10) or “executioners” of apoptosis (eg, CASP3, CASP6, and CASP7). Initiator procaspases contain large prodomains, whereas executioner procaspases contain small prodomains.
Hundreds of caspase substrates have been identified, including cytoskeleton proteins (eg, Actin and Gelsolin), nuclear proteins (eg, Lamin A and B), proteins involved in DNA damage repair (eg, PARP, RAD51, and DNA-PKcs), cell-cycle proteins (eg, p21, p27, CDC27, and RB), cytokines (eg, IL-1β and IL-18) and apoptotic proteins (eg, Caspases, BCL-2, BCL-XL, BID, BAX, and ICAD) (Luthi and Martin, 2007). The caspase-activated DNase (CAD) is inactive when associated with its inhibitor ICAD (also known as DNA fragmentation factor [DFF]). In response to apoptotic stimuli, ICAD is cleaved by caspases allowing the release of the active endonuclease CAD to cleave DNA of apoptotic cells to produce internucleosomal DNA cleavage, a characteristic of apoptosis.
A number of caspases also have proinflammatory functions. These caspases include CASP1, -4, -5, -11, and -12 (Li and Yuan, 2008). CASP8 is important in other cellular functions, including blood vessel formation during embryogenesis and mitogen- or antigen-induced proliferation of T and B cells (Su and Lenardo, 2008).
The hierarchical involvement of caspases in the extrinsic and intrinsic apoptotic pathways, impaired apoptosis in their absence, and the nonapoptotic functions ascribed for the caspases all highlight the importance of this family of proteins.
9.4.2.3 Cytochrome c, APAF-1, and Apoptosis The apoptosis protease activating factor-1 (APAF-1) is the first identified mammalian ortholog of Ced-4. Studies of APAF-1 have demonstrated its critical role for the intrinsic apoptotic pathway (Tait and Green, 2010). APAF-1 is a 130-kDa protein composed of 3 functional domains: a short N-terminal CARD, a central Ced-4 homology domain, and a long C-terminal “WD-40” repeat domain. Following the release of Cytochrome C from mitochondrial intermembrane space, it binds to APAF-1, and induces its conformational change and oligomerization, thus leading to the formation of the apoptosome (see Fig. 9–12). Similar to the death-inducing signaling complex (DISC) and CASP8, the apoptosome recruits, dimerizes, and activates the initiator CASP9, thus leading to apoptosis. A number of studies have demonstrated that Cytochrome C, APAF-1, and CASP9 are all required for the activation of the downstream effectors CASP3, -6, and -7. Transcriptional regulation of APAF-1 has been reported to be mediated by E2F1 and p53, suggesting its possible contribution to E2F1- and p53-mediated apoptosis. The BCL-2 family member and antiapoptotic protein BCL-XL interacts with APAF-1 and CASP9, and forms a ternary complex. Through this interaction, BCL-XL negatively regulates CASP9 activation.
By mediating apoptosome formation, Cytochrome C and APAF-1 play essential roles in the mitochondrial apoptotic pathway.
9.4.3 The Mitochondrial Apoptotic Pathway
Apoptotic stimuli, which trigger the mitochondrial or intrinsic apoptotic pathway, activate BAX and BAK and lead to mitochondrial outer membrane permeabilization and the release of a number of proteins important for apoptosis (eg, Cytochrome C, SMAC, and OMI) from the mitochondrial intermembrane space into the cytosol (see Fig. 9–12; Tait and Green, 2010). Mitochondrial membrane permeabilization is suppressed by the antiapoptotic BCL-2 family members (eg, BCL-2 and BCL-XL), as they bind and inhibit activated BAX or BAK as well as other BH3-only proteins. Once in the cytosol, Cytochrome C forms the apoptosome with APAF-1 and CASP9, thus leading to activation of CASP9, which, in turn, processes and activates downstream effector caspases. This leads to cleavage of various cellular substrates, and cell death.
Caspase activity is modulated by various members of the family of inhibitors of apoptosis (IAPs). IAPs were originally discovered in baculoviruses and shown to suppress the host cell death response to viral infection (Altieri, 2010). Currently, 8 human IAPs have been identified, including the X-linked IAP (XIAP), Survivin, and cellular IAP1 and -2 (c-IAP1 and c-IAP2). Several IAPs bind directly to caspases such as CASP3, -7, and -9 and inhibit their functions. This IAPs-mediated inhibition of caspases can be antagonized by the proteins SMAC and OMI (see Fig. 9–12). When SMAC and OMI are released from the mitochondrial intermembrane space into the cytosol, they bind directly to XIAP and suppress its inhibition of caspases. In addition to their roles in apoptosis, IAPs are also involved in other processes including nuclear factor kappa B (NF-kB) signaling, cell division, and cellular stress responses.
The mitochondrial apoptotic pathway is essential for the response to DNA damage and oncogenic activation. The tumor suppressor p53 plays a critical role in this apoptotic pathway as it controls the transactivation of a number of essential proapoptotic BCL-2 family members including PUMA, BAX, and NOXA. In addition to controlling different cellular processes, including proliferation (see Sec. 9.4.1), the activated oncogene c-MYC (see Chap. 7, Sec. 7.5.2) also induces apoptosis (Meyer and Penn, 2008). c-MYC induced apoptosis is p53 dependent (see Fig. 9–12). Deregulated c-MYC also promotes the accumulation of the tumor suppressor ARF that sequesters MDM2 in the nucleolus, thus releasing p53 from MDM2-mediated inhibition. Thus the ARF-MDM2-p53 pathway is important for c-MYC–induced apoptosis.
Posttranslational modifications play important roles in the regulation of the mitochondrial apoptotic pathway (Newton and Vucic, 2007). For example, MCL-1 is ubiquitinated by ARF-BP1, targeting it for proteasomal degradation (Fig. 9–4B). Evidence exists that IAPs ubiquitinate mammalian caspases and the Drosophila IAP (DIAP1) polyubiquitinates the caspase orthologs Dronc, Dcp-1, and drICE leading to their nondegradative inactivation. Polyubiquitination of BCL-2, BAX, BIK, NOXA, BID, and BIM has also been reported to lead to their proteasomal degradation, thus controlling their levels of expression. Deregulation of this apoptotic pathway has been associated with a number of diseases, including cancer.
9.4.4 The Death Receptor Apoptotic Pathway
The death receptor or extrinsic apoptotic pathway is initiated by the interaction of the death receptors, members of the tumor necrosis factor (TNF) receptor superfamily, with their ligands (Wilson et al, 2009). The death receptors share the presence of an approximately 80-amino-acid motif known as the death domain (DD) in their cytoplasmic tails. Six human death receptors have been identified and include FAS (also known as CD95 or APO-1), TNFR-1, death receptor (DR) 3, DR4 (also known as TRAILR1), DR5 (also known as TRAILR2), and DR6. Several ligands for these death receptors have been identified and include TNF and LTα (TNFR1), FASL (FAS), TL1A (DR3), and APO2L/TRAIL (DR4 and DR5). The ligand for DR6 remains unknown.
The death receptor apoptotic pathway is exemplified by the FAS-induced apoptotic signaling pathway (see Fig. 9–12; Strasser et al, 2009). Following FAS/FASL interaction, the FAS receptor proteins aggregate to form a trimer and recruit the adaptor protein FADD (FAS-associated death domain) that contains 2 protein interaction domains, a death domain, and a death effector domain (DED). FAS/FADD interaction allows the recruitment of the DED-containing initiator pro-CASP8 (and also pro-CASP10 in human) and the formation of the DISC. Dimerization of pro-CASP8 within the DISC allows its autoprocessing, activation and the formation and release of its active tetrameric form into the cytosol. Active CASP8 in the cytosol processes its substrates including the effector CASP3, CASP6 and CASP7, and the BH3-only protein BID. Cellular FLICE-like inhibitory protein (cFLIP), a pseudo-CASP8 protein with a nonfunctional catalytic domain, inhibits the death receptor apoptotic pathway by precluding the recruitment of CASP8 to the DISC.
p53 also affects the death receptor apoptotic pathway as demonstrated by p53 transactivation of FAS and DR5 in response to DNA damage (see Figs. 9–12 and 9–15). Posttranslational modifications, including ubiquitination, also play important roles in the death receptor apoptotic pathway (Wertz and Dixit, 2010). In response to stimulation of DR4 or DR5, the E3 ligase CULLIN-3 polyubiquitinates CASP8 present at the DISC. This ubiquitination event recruits the ubiquitin-binding polypeptide p62 that mediates CASP8 aggregation, thus increasing its activation and processing (Jin et al, 2009). c-FLIP, an inhibitor of CASP8 and CASP10, is transcriptionally upregulated by NF-κB and is also regulated by ubiquitylation. The E3 ligase ITCH has been shown to mediate the polyubiquitylation of c-FLIP, leading to its proteasomal degradation (Chang et al, 2006).
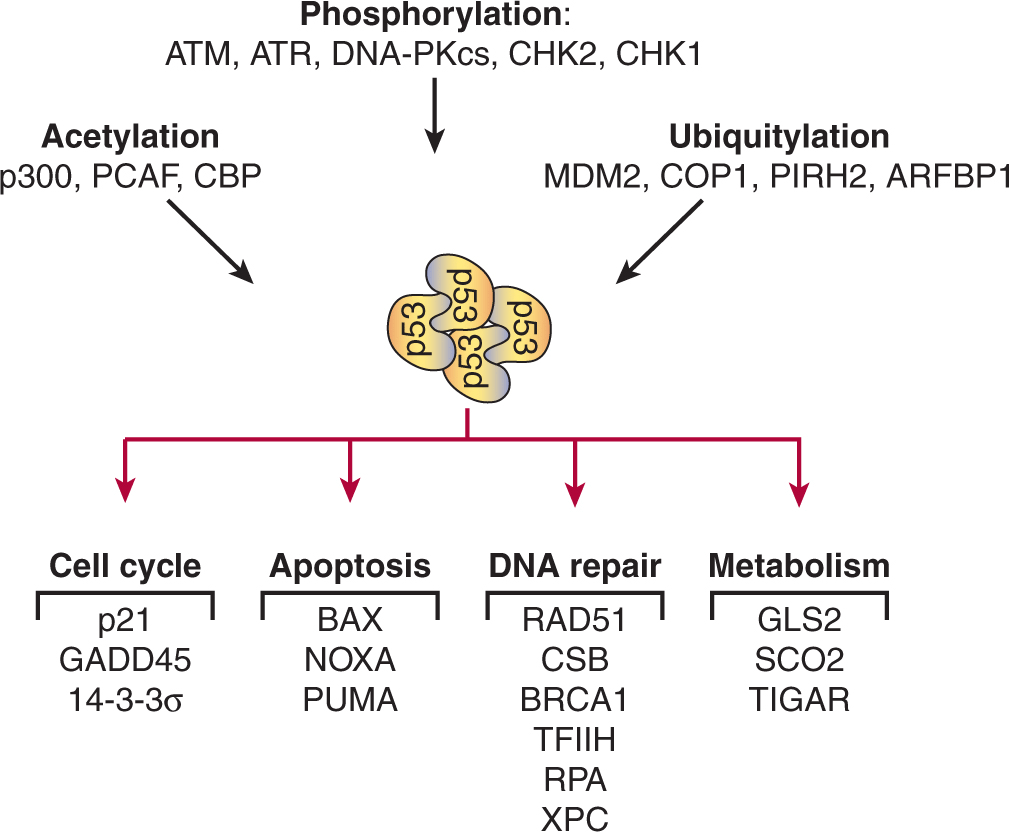
FIGURE 9–15 The tumor suppressor p53 is a mutlifunctional protein highly regulated by posttranslational modifications. Examples of proteins involved in p53 posttranslational modifications (eg, acetylation, phosphorylation, and ubiquitylation) are indicated. Activated p53 plays major roles in a number of cellular processes including cell cycle, apoptosis, DNA repair, and metabolism. Examples of p53 transcriptional targets involved in these cellular processes are indicated (see also Chap. 7, Sec. 7.6.1).
Communication between the death receptor and mitochondrial apoptotic pathways is best demonstrated by CASP8 cleavage of BID. This cleavage generates a truncated form of BID (tBID) that cooperates with BAX to form openings in the outer mitochondrial membrane, leading to release of proteins including Cytochrome C from the mitochondrial intermembrane space (see Fig. 9–12). This CASP8 processing of BID amplifies the death receptor apoptotic pathway. In addition, stimulation of the mitochondrial pathway leads to the activation of the effector caspases, such as CASP3 and CASP6, that can subsequently process and activate CASP8.
Impaired apoptosis owing to mutations in the components of the death receptor apoptotic pathway has been observed in human and mouse cells. For example, resistance to stimuli that activate this pathway was observed in cells deficient for CASP8, FADD, FAS, FASL, or in cells overexpressing c-FLIP (Strasser et al, 2009). Similar to the mitochondrial apoptotic pathway, the death receptor apoptotic pathway also plays critical roles in development and tissue homeostasis, and is dys-regulated in a number of diseases including cancer.
9.4.5 Apoptosis, Developmental Defects, and Disease
Deregulation of apoptosis is associated with human diseases, including immunodeficiency, neurodegeneration, and auto-immunity. Similarly, studies of genetically modified mice deficient in or overexpressing anti- or proapoptotic proteins demonstrate an essential role of apoptosis in the prevention of embryonic and postnatal developmental defects, and to restrain the development of various diseases, including autoimmunity.
9.4.5.1 Antiapoptotic BCL-2 Family Members and Disease BCL-2 inhibits apoptosis in response to a wide range of stimuli that target the mitochondrial apoptotic pathway. In addition to cancer, overexpression of the prosurvival BCL-2 family members also promotes the development of other diseases, including autoimmunity. Transgenic mice expressing the human BCL-2 in T and B cells develop an autoimmune disorder characterized by antinuclear antibodies and glomerulonephritis (Strasser et al, 1991).
Deficiency of the prosurvival BCL-2 family members (eg, BCL-2, BCL-XL or MCL-1) results in increased cell death and promotes the development of a number of pathologies (Youle and Strasser, 2008). Mice deficient for BCL-2 display polycystic kidney disease and loss of mature B and T cells as a consequence of increased apoptosis. Bcl-2–/– mice die by 6 weeks of age. Inactivation of Bcl-XL in mice results in embryonic lethality by day 14 of gestation, which underscores the important role of BCL-XL in the regulation of programmed cell death during the development of embryonic nervous system and lymphoid organs. Similarly, analysis of mice deficient for MCL-1 demonstrated the importance of this antiapoptotic protein for the survival and implantation of the zygote, while conditional gene targeting of Mcl-1 results in the premature death of immature and mature B and T cells and hematopoietic stem cells (Opferman et al, 2003).
9.4.5.2 Proapoptotic BCL-2 Family Members and Disease The proapoptotic members of the BCL-2 family also play critical roles in apoptosis and their deregulation has been associated with various pathologies (Youle and Strasser, 2008). While Bax–/– mice display male sterility and mild lymphoid hyperplasia, Bak–/– mice display no such abnormalities. However, perinatal lethality was observed in Bax–/–Bak–/– mice. The small subset of double mutant mice that survive to adulthood accumulate lymphoid and myeloid cells and typically develop lymphoadenopathy and splenomegaly. The surviving Bax–/–Bak–/– mice also accumulate cells of the central nervous system and fail to remove their interdigital webs.
Other mouse models deficient for 1 or more BH3-only proteins have been generated (Youle and Strasser, 2008). For example, Bid–/– mice display hepatocyte resistance to FAS-induced apoptosis and suffer fatal hepatitis. Mice deficient for either BIM or PUMA display resistance to various apoptotic stimuli that target the intrinsic apoptotic pathway. Similar to Bax–/– Bak–/– mice, mice lacking both BIM and PUMA displayed higher resistance to apoptosis compared to single mutants, thus indicating cooperation between these 2 proteins. Puma–/– mice possess a normal life span and show no increased predisposition to diseases. In contrast, because of its important roles in the deletion of autoreactive T and B cells and its requirement for the downregulation of immune responses, Bim–/– mice display splenomegaly and lymphadenopathy, and in a C57BL/6x129SV genetic background they develop a fatal systemic lupus erythematosus (SLE)-like autoimmune disease. Recent studies demonstrate that the autoimmunity of Bim–/– mice is further exacerbated in a Fas mutant background (Strasser et al, 2009). These studies demonstrate the collaboration of the mitochondrial and the death receptor apoptotic pathways in the elimination of autoreactive cells and the prevention of autoimmune disorders.
Collectively, the data strongly support that both the anti-and the pro-apoptotic BCL-2 family members are important for in vivo apoptosis, and that deregulation of a number of proteins in this family leads to developmental defects and immune disorders.
9.4.5.3 Caspases and Disease Mice deficient for specific caspases demonstrate the crucial role for apoptosis in development (Li and Yuan, 2008). Casp3–/– and casp9–/– mice exhibit a similar brain defect characterized by ectopic masses of supernumerary cells that escape apoptosis during brain development (Fig. 9–16; Hakem et al, 1998; Woo et al, 1998). Casp12–/– mice are resistant to apoptosis induced in response to stress to the endoplasmic reticulum, but undergo apoptosis in response to other stimuli (eg, FAS or dexamethasone). Casp2–/– mice are viable, and although most of casp2–/– cells fail to show any significant defect in apoptosis, casp2–/– B lymphoblasts display defective cytolysis mediated by Perforin and Granzyme B. Casp2–/– females display excess ovarian germ cells and resistance of oocytes to antineoplastic drug-induced apoptosis.
FIGURE 9–16 Developmental defects associated with deficiency of caspases. Postnatal day 1 wild-type, Caspase 3 deficient (casp3–/–), and Caspase 9 deficient (casp9–/–) mice are shown. Arrows point to the characteristic perturbation of brain (cortex and forebrain) structures observed in both casp3–/– and casp9–/– mutants.
The autoimmune lymphoproliferative syndrome (ALPS), a rare inherited human disorder, is characterized by lymphoproliferation, accumulation of CD4–CD8– (double-negative) T lymphocytes and autoantibody production (Su and Lenardo, 2008). This syndrome is associated with heterozygous mutations of FAS or FASL. ALPS patients are at an increased risk to develop lymphomas. Mice that carry homo-zygous mutations of FAS or FASL also develop an ALPS-like syndrome characterized by splenomegaly, lymphadenopathy, hypergammaglobulinemia, autoantibody production, and glomerulonephritis. Homozygous mutations of CASP8 are associated with an ALPS-like syndrome. Patients with this rare syndrome are immunodeficient and display lymphadenopathy, splenomegaly, and impaired lymphocyte activation. Casp8–/– mice display defects in the heart and neural tube, accumulate erythrocytes, and die by embryonic day 12 (Li and Yuan, 2008). Remarkably, similar to the ALPS-like syndrome of CASP8 mutant patients, mice with specific inactivation of casp8 in T-cell lineage are also immunodeficient, exhibit impaired T-cell homeostasis characterized by T-cell lymphopenia, and have impaired responses to viral infection (Salmena et al, 2003). These mice also have short lifespans as they develop an age-dependent and fatal lymphoproliferative disorder (Salmena and Hakem, 2005). In addition, CASP8 deficiency in the B-cell lineage promoted development of lymphomas in mouse models (Hakem et al., 2012).
Caspases are therefore important for in vivo apoptosis. Impairment of their functions can lead to defective embryonic and postnatal development, and promote the development of diseases including immunodeficiency and lymphoproliferation.
9.4.6 Mechanisms Leading to Necrosis
Although necrosis was thought initially to be a nonregulated process, recent studies demonstrate that necroptosis, a programmed type of necrosis, is highly regulated by a network of proteins (Christofferson and Yuan, 2010). Ligands binding to death receptors upon the plasma membrane of normal cells leads to CASP8-dependent cleavage and inactivation of the receptor-interacting protein RIPK1 and RIPK3 (also known as RIP1 and RIP3, respectively) (Fig. 9–17). Activated CASP8 also cleaves downstream targets including the executioner Caspases 3, 6, and 7, and thus promotes cell death by apoptosis. However, together with the presence of defective death receptor signaling, cells lacking CASP8 activity fail to induce cleavage of RIPK1 and RIPK3. These kinases are therefore normally active in CASP8 deficient cells and can trigger cell death by necroptosis. T cells deficient for CASP8 die by necroptosis and inhibitors of necroptosis (eg, necrostatin 1) rescue the survival of these T cells. A genome wide short-interference RNA screen for regulators of necrosis has identified 432 genes that regulate this type of necrotic cell death (Hitomi et al, 2008). BMF, a BH3-only BCL2 family member was found to be important for death receptor-induced necrosis. Recent data demonstrated that genetic inactivation of Ripk3 rescues embryonic lethality of casp8–/– mice and restores survival of their T cells (Peter, 2011). Similarly, the absence of Ripk1 rescued the embryonic lethality of Fadd–/– mice and the proliferative defects of their T cells.
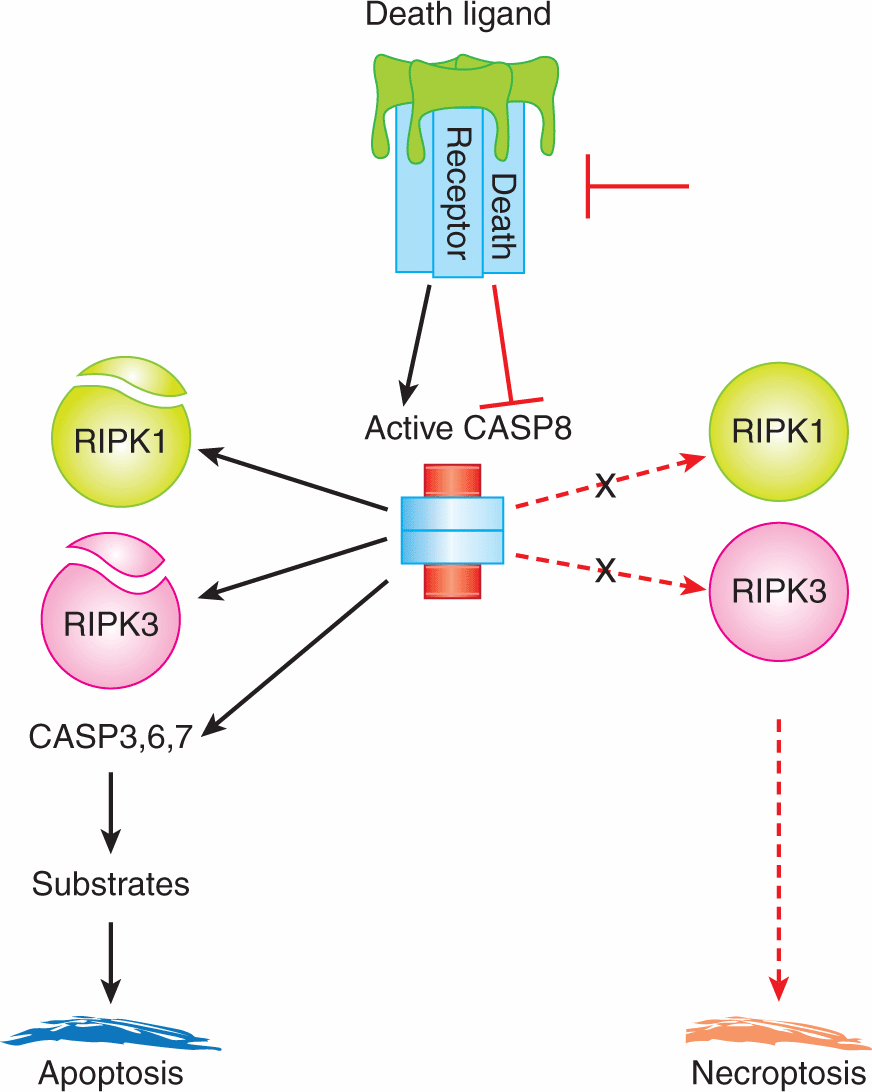
FIGURE 9–17 Representation of the crosstalk between apoptosis and necroptosis. In response to the binding of ligands to death receptors (eg, TNFR1) on the plasma membrane, active CASP8 inactivates RIPK1 and RIPK3 by proteolytic cleavage and also processes downstream caspases to promote apoptosis. In the case where death receptor signaling is impaired or CASP8 is lost or inhibited, the executioner caspases are not activated and RIPK1 and RIPK3 are not cleaved and thus remain active. Active RIPK1 and RIPK3 trigger necroptosis, a programmed necrotic cell death.
These genetic data demonstrate the strong interactions between the mechanisms that control apoptotic and necrotic cell death.
9.4.7 Modifications to Control of Cell Death in Cancer
Resisting programmed cell death has been proposed as 1 of 6 hallmarks of cancer (Hanahan and Weinberg, 2000). A number of tumor suppressors (eg, p53) and oncogenes (eg, c-MYC, BCL-2; see Chap. 7, Sec. 7.5.2) important for human cancer are involved in the regulation of apoptosis. The p53 gene, important for promoting apoptosis and suppressing proliferation, is mutated in more than 50% of human tumors. Consistent with the critical role p53 plays in suppressing human cancer, mouse models for p53 deficiency have increased predisposition for various types of tumors (Lozano, 2010).
p53 is required for the apoptotic response to various stimuli, including DNA damage, oncogenic activation, hypoxia, and ribosomal stress. As a transcription factor, p53 controls the expression of genes involved in different cellular processes such as apoptosis, proliferation, metabolism, and DNA repair (see Fig. 9–15). Apoptotic genes that are transcriptionally activated by p53 include Bax, Noxa and Puma.
The Bcl-2 gene, was identified initially in studies of human follicular B-cell lymphoma carrying the translocation t(14-18). This chromosomal translocation results in the overexpression of BCL-2 and inhibition of apoptosis. The antiapoptotic protein MCL-1 is also overexpressed in a number of human malignancies including B-cell lymphoma, and myeloma (Warr and Shore, 2008). Mutations of the proapoptotic proteins are also observed in human cancers. Thus, human Bax was reported to be mutated in colon cancer and hematopoietic malignancies (Meijerink et al, 1998; Rampino et al, 1997). Loss of CASP8 expression because of hypermethylation of its promoter has been observed in human neuroblastomas with N-MYC amplification, small cell lung carcinoma, and in relapsed glioblastoma multiforme. Inactivation of human Casp8 by somatic mutations is observed in hepatocellular carcinomas and advanced gastric cancer (Fulda, 2009). In addition to cancer development, loss of CASP8 is also reported to promote metastasis of neuroblastoma.
Studies of mouse models have confirmed the important role apoptosis plays in suppressing cancer. Transgenic mice overexpressing BCL-2 in lymphocytes develop lymphomas (McDonnell and Korsmeyer, 1991) and risk for lymphoma was also increased in transgenic mice overexpressing MCL-1 in hematopoietic and lymphoid tissues (Zhou et al, 2001). BCL-2 inhibits c-MYC–induced apoptosis and, consequently, transgenic mice overexpressing both BCL-2 and c-MYC in B-cell lineages (Strasser et al, 1990) or mammary glands (Jager et al, 1997) display elevated cancer risk compared to c-Myc transgenic mice. c-MYC–induced tumorigenesis is also promoted by the overexpression of the antiapoptotic proteins BCL-XL and MCL-1 (Campbell et al, 2010; Cheung et al, 2004).
Radiation and most chemotherapeutic agents (eg, platinum derivatives) used in the clinic trigger the death of tumors. There is a continous effort to generate novel compounds to modulate cell death, particularly apoptosis, and improve the response of cancer patients to therapies (Ashkenazi, 2008 #991; Davids, 2012 #990). Some of these compounds are currently in clinical trials as single agents or in combination with other chemotherapeutic drugs. Compounds that activate the death receptor pathway include agonists and human monoclonal antibodies to proapoptotic receptors DR4 and DR5 and their ligand APO2L/TRAIL. Radiation and most chemotherapeutic agents kill through activation of the mitochondrial apoptotic pathway; therefore, significant efforts have been made to identify compounds to activate this pathway. Approaches have been designed to inhibit the prosurvival members of BCL-2 family or activate its pro-apoptotic members. This is exemplified by the small molecule BH3 mimetic ABT-737, and the closely related and orally bioavailable ABT-263. These compounds bind to BCL-2, BCL-X, and BCL-W and kill tumors by displacing pro-apoptotic proteins from BCL-2. Promising results have been obtained with ABT-263, and several clinical trials are underway to assess therapeutic benefit of its combination with standard anti-cancer agents.
In addition to promoting development of a number of diseases, including neurodegenerative diseases and immune disorders, defective apoptosis also promotes cancer development. Mechanisms that lead to cell death or survival can be targeted to improve cancer therapy.
Related posts:
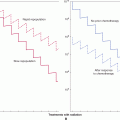
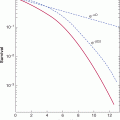
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


