Cell Cycle
Steven I. Reed
Cell division is a process that must be carried out with absolute fidelity. The program of generating an adult organism from a single zygote involves countless cell duplications, each requiring the precise partitioning of genetic material and most other cellular components to daughter cells. The division process then continues during adult life to replenish essential cells restricted to a limited lifespan. As a result, organisms have evolved cell-duplication strategies that include redundant safeguards to prevent errors or, if errors occur, to correct them. Nevertheless, errors do occur at a measurable frequency, and mutations accumulated over time can weaken protective mechanisms, rendering the genome increasingly vulnerable to challenges. The resulting loss of genetic and genomic stability has serious implications for survival in that it is a major contributing factor to the development of malignancy. Indeed, cancer is one of the leading causes of mortality in humans.
In this chapter, the basic principles of mammalian cell division and the mechanisms that have evolved to safeguard the integrity of the process are reviewed. Then there is a discussion of how the normal control mechanisms of cell division and protective safeguards become subverted in cancer cells. It is hoped that, ultimately, detailed knowledge of cell division in normal and cancer cells will lead to rational effective therapeutic approaches.
CELL-CYCLE ENGINE
Although the details of cell division vary considerably across phylogenetic lines and even in different cell types within the same organism, the underlying infrastructure that mediates and controls the cell division process is remarkably conserved. If one compares yeast cells and mammalian cells in culture, perhaps the two most aggressively studied model cell division systems are not only the respective cell division cycles organized along a similar scheme, but many of the proteins used in the cell division pathway are easily recognizable as being evolutionarily related. Indeed, some of these proteins are so highly conserved that they are functional in the heterologous organism despite a billion years of divergent evolution.
Phases of the Cell Cycle
As alluded to previously, the basic organization of the cell cycle is highly conserved in eukaryotic evolution. In 1951, Howard and Pelc,1 studying the division of plant root cells, separated the process into four phases eventually referred to as GAP1, synthetic phase, GAP2, and mitosis. The shorthand that emerged from this descriptive work (G1, S phase, G2, and M phase or mitosis) has been the lens through which all subsequent dividing cells have been observed, and the four successive phases are referred to collectively as the cell cycle. The key observation made by these investigators was that the events that together make up the cell division process do not all occur continuously. Specifically, although growth and protein synthesis occur constantly for the most part, synthesis of DNA occurs only during a discrete interval. The preceding phase was designated GAP1 or G1, and the subsequent phase before cell division was referred to as GAP2 or G2. Although at the time little could be said concerning what a cell did during these silent “gap” phases, it is now known that these are not idle periods in a cell’s life but the intervals in which most regulation of the cell cycle is specifically exerted. A large amount of information, originating from the external environment and the cell’s internal milieu, is integrated during the G1 and G2 intervals and used to determine whether and when to proceed into S phase and M phase, respectively.
Mitosis, the most visibly dynamic interval of the cell cycle, has itself been traditionally subdivided into five phases: prophase, prometaphase, metaphase, anaphase, and telophase. In metazoans and plants (as opposed to fungi) mitosis entails a particularly dramatic change of state for the cell. During prophase most of the internal membranous compartments of the cell, including the nucleus, are disassembled and dispersed. Replicated chromosomes (chromatids) are condensed into paired compact rods, and a bipolar microtubule spindle is assembled. Biosynthesis of proteins (transcription and translation) largely ceases. During prometaphase, chromosomes form bivalent attachments to the spindle, driving them to the cellular equator. Proper alignment of paired chromatids on the spindle is indicative of metaphase. During anaphase, the paired sister chromatids lose cohesion and microtubule forces
separate the chromatids and pull them to opposite poles of the cell. During telophase, the events of prophase are reversed: The nuclei and other membrane structures reassemble, the chromosomes decondense, and protein synthesis resumes. After mitosis, the two daughter cells pull apart and separate in a process known as cytokinesis. Current knowledge of the cell cycle has accrued historically from a number of different experimental approaches and systems. In the early 1970s, experiments carried out by fusing mammalian cells in different cell-cycle phases revealed the existence of dominant inductive activities for the S phase and the M phase.2,3 Shortly thereafter, similar inductive activities were isolated from mature frog eggs arrested at meiotic metaphase II and shown to be capable of inducing G2 oocytes to enter into meiotic divisions,4 equivalent in many respects to mitosis. At the same time, genetic analysis of cell division in yeast revealed that the products of individual genes controlled specific events in the cell cycle and that these events could be organized in pathways, much like metabolic pathways.5 Eventually, all of these lines of investigation converged in the 1980s, leading to the discovery of cyclin-dependent kinases (CDKs).
separate the chromatids and pull them to opposite poles of the cell. During telophase, the events of prophase are reversed: The nuclei and other membrane structures reassemble, the chromosomes decondense, and protein synthesis resumes. After mitosis, the two daughter cells pull apart and separate in a process known as cytokinesis. Current knowledge of the cell cycle has accrued historically from a number of different experimental approaches and systems. In the early 1970s, experiments carried out by fusing mammalian cells in different cell-cycle phases revealed the existence of dominant inductive activities for the S phase and the M phase.2,3 Shortly thereafter, similar inductive activities were isolated from mature frog eggs arrested at meiotic metaphase II and shown to be capable of inducing G2 oocytes to enter into meiotic divisions,4 equivalent in many respects to mitosis. At the same time, genetic analysis of cell division in yeast revealed that the products of individual genes controlled specific events in the cell cycle and that these events could be organized in pathways, much like metabolic pathways.5 Eventually, all of these lines of investigation converged in the 1980s, leading to the discovery of cyclin-dependent kinases (CDKs).
Cyclin-Dependent Kinases
Arguably the most significant advance in understanding cell-cycle regulation was the discovery of CDKs.6 These are binary, proline-directed, serine-threonine -specific protein kinases that consist of a catalytic subunit (the CDK) that has little if any intrinsic enzymatic activity and a requisite positive regulatory subunit known as a cyclin. In yeast, one CDK and numerous cyclins carry out cell-cycle regulatory functions, whereas in mammals, these same functions are carried out by a number of different CDKs and cyclins. In yeast, in which multiple cyclins activate the same CDK (CDK1, also known as Cdc28 or Cdc2, depending on the species) for distinct cell-cycle tasks, it is clear that most if not all substrate specificity beyond a rather degenerate primary structure target consensus lies in the cyclin subunit. In mammals, it is likely that substrate specificity is shared by CDK and cyclin subunits. Although not all pairwise combinations are permitted, there are enough combinatorial possibilities to create a significant level of substrate specificity.
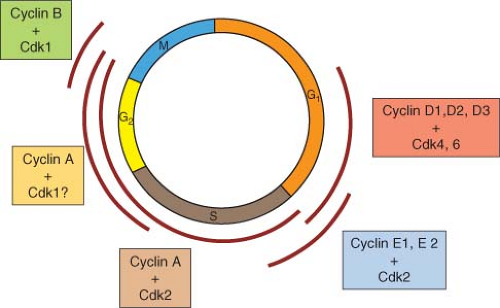 FIGURE 6.1 Windows of cyclin-dependent kinase (CDK) function in the cell cycle. D-type cyclins (cyclins D1, D2, and D3) activate CDK4 and CDK6 for functions extending from middle G1 to the G1/S-phase transition. E-type cyclins (cyclins E1 and E2) activate CDK2 for functions at the G1/S-phase boundary, probably extending into early S phase. Cyclin A activates CDK2 for functions extending from the G1/S-phase boundary and extending into G2. Cyclin A is known to interact with CDK1 as well; however, no specific function for this complex has been identified. Finally, cyclin B activates CDK1 at the G2/M-phase boundary with activity that lasts until cyclin B is degraded during anaphase. |
CDKs have a structure similar to that of other protein kinases, consisting of two globular domains (the N-lobe and C-lobe) held together by a semi flexible hinge region. Protein substrates bound by the active enzyme are thought to fit into a cleft between the two domains. The N-lobe contains the adenosine triphosphate (ATP)-binding site. Studies comparing CDKs and CDK-cyclin complexes based on x-ray diffraction crystallography indicate that the primary role of the cyclin, in addition to substrate docking functions, is to realign critical active site residues into a catalytically permissive configuration and to open the catalytic cleft to accommodate substrates.7 Once bound to a cyclin, the CDK active site is configured similarly to other protein kinases that do not require cyclin binding.
The known CDKs and cyclins and their presumptive intervals of function in the mammalian cell cycle are summarized in Figure 6.1. For the
most part, the functional intervals of CDKs are determined by the accumulation and disappearance of cyclins. Whereas CDKs tend to be expressed at a constant level through the cell cycle, cyclin accumulation is usually dynamic, regulated at the level of biosynthesis and degradation (discussed in greater detail in “Cell-Cycle Machinery”). To summarize, three partially redundant D-type cyclins (D1, D2, and D3) activate two partially redundant CDKs (CDK4 and CDK6). Although, unlike most other cyclins, D-type cyclins do not appear to be expressed with high periodicity in cycling cells, the interval at which their primary activating function is thought to occur is from mid-to-late G1 to direct phosphorylation of the cell-cycle inhibitor pRb and related proteins p107 and p130. Phosphorylation of these proteins by cyclin D-CDK4/6 inactivates their negative regulatory functions, allowing progression into S phase.8,9
most part, the functional intervals of CDKs are determined by the accumulation and disappearance of cyclins. Whereas CDKs tend to be expressed at a constant level through the cell cycle, cyclin accumulation is usually dynamic, regulated at the level of biosynthesis and degradation (discussed in greater detail in “Cell-Cycle Machinery”). To summarize, three partially redundant D-type cyclins (D1, D2, and D3) activate two partially redundant CDKs (CDK4 and CDK6). Although, unlike most other cyclins, D-type cyclins do not appear to be expressed with high periodicity in cycling cells, the interval at which their primary activating function is thought to occur is from mid-to-late G1 to direct phosphorylation of the cell-cycle inhibitor pRb and related proteins p107 and p130. Phosphorylation of these proteins by cyclin D-CDK4/6 inactivates their negative regulatory functions, allowing progression into S phase.8,9
Unlike D-type cyclins, E-type cyclins (E1 and E2) are expressed with high cell-cycle periodicity, accumulating in late G1 and declining during S phase. E-type cyclins activate CDK2, and the fact that premature expression of cyclin E1 leads to accelerated entry into S phase10,11 has suggested that the target(s) must be proteins responsible for initiation of DNA replication. However, the essentiality of cyclin E-CDK2 in this context has been put into question by the demonstration that cells from cyclin E1/E2 nullizygous mouse embryos can cycle with reasonably normal kinetics and can certainly initiate DNA replication.12 The most likely explanation for the dispensability of E-type cyclins for S phase functions is redundancy with cyclin A, which also activates CDK2.
Cyclin A accumulates initially at the G1/S-phase boundary and persists until prometaphase of mitosis. It has been best characterized as an activator of CDK2; however, it has also been reported to form complexes with CDK1. It is presumed that CDK2, activated by E-type cyclins and cyclin A, promotes cell-cycle progression from the G1/S boundary through the G2 interval.
At this time, B-type cyclins, in conjunction with CDK1, are responsible for getting cells into and through mitosis. Although mammalian cells express a number of B-type cyclins, only cyclin B1 appears to be essential. Cyclin B1 accumulates through S phase and G2 and then is degraded at the metaphase-anaphase transition. It should be pointed out that the CDK family is extensive and that eukaryotes possess many additional CDKs that ostensibly have nothing to do with cell-cycle regulation.
The blueprint for CDK function through the mammalian cell cycle presented here is based on a large body of experimental evidence and most likely accounts for primary activities at each cellcycle stage. However, this model does not account for potential redundancy of CDK function. Recently, however, it has been demonstrated that a CDK2-nullizygous mouse is viable, and furthermore, relatively normal.13,14 Investigation of embryonic fibroblasts from these mice has suggested that in the absence of CDK2, CDK1 can carry out the functions normally attributed to CDK2.15 However, the contribution of CDK1 to these functions in unperturbed cells remains to be determined.
Modes of Cyclin-Dependent Kinase Regulation
Because the activity of CDKs is central to cell survival, these enzymes are, of necessity, highly regulated.6 As a result, a number of diverse regulatory mechanisms have evolved to allow for integration of environmental and internal signals (Fig. 6.2). A primary mode of CDK regulation is the availability of activating cyclins, as alluded to previously. For most cell-cycle regulatory CDKs, the relevant cyclins exhibit a distinct temporal program of accumulation and degradation, determining a precise window of CDK activation. Although D-type cyclins tend not to be highly regulated in cycling cells, they are strongly down-regulated as cells exit the cell cycle into a nonproliferative state and then resynthesized in response to mitogen stimulation and cell-cycle re-entry. The genes encoding cyclin E1 and E2 are transcribed periodically late in G1 and up to the G1-S phase transition. This, coupled with ubiquitin-mediated proteolysis of cyclin E in active cyclin E-CDK2 complexes, creates the observed window of cyclin E accumulation from late G1 to mid-S phase.
Like cyclin E, the accumulation of cyclin A is determined by periodic transcription. However, unlike cyclin E, cyclin A remains stable in active CDK2 complexes. The timing of ubiquitinmediated proteolysis of cyclin A is determined by activation of a protein-ubiquitin ligase known as the anaphase-promoting complex/cyclosome (APC/C) in prometaphase. Thus, the window of cyclin A accumulation is from the G1-S transition until early in mitosis. Finally, B-type cyclin accumulation is also linked to periodic transcription. In this case, transcription begins in late S phase and persists through G2. Similarly to cyclin A, B-type cyclins are targeted for ubiquitin-mediated proteolysis by the APC/C during mitosis, although their disappearance occurs slightly later in mitosis than that of cyclin A.
It is interesting to note that periodic transcription of cyclins E, A, and B mRNAs relies primarily on negative regulation. For cyclin E, an element known as CERM (cyclin E repressor module) binds a repressor complex containing the repressive member of the E2F family of transcription factors, E2F4, as well as the Rb-related protein p107 and a histone deacetylase.16 Inactivation of the repressive complex in late G1 via phosphorylation of p107
by CDK4/6 allows the constitutive transcription factor, Sp1, to drive cyclin E transcription. Transcription of cyclin A and B mRNAs is similarly regulated. In this case, the repressor element is known as cell cycle genes homology region (CHR)17 but the corresponding repressor complex has not yet been well characterized. However, once repression is relieved, transcription of both cyclin A and B mRNAs is driven the constitutive transcription factor, NF-Y.
by CDK4/6 allows the constitutive transcription factor, Sp1, to drive cyclin E transcription. Transcription of cyclin A and B mRNAs is similarly regulated. In this case, the repressor element is known as cell cycle genes homology region (CHR)17 but the corresponding repressor complex has not yet been well characterized. However, once repression is relieved, transcription of both cyclin A and B mRNAs is driven the constitutive transcription factor, NF-Y.
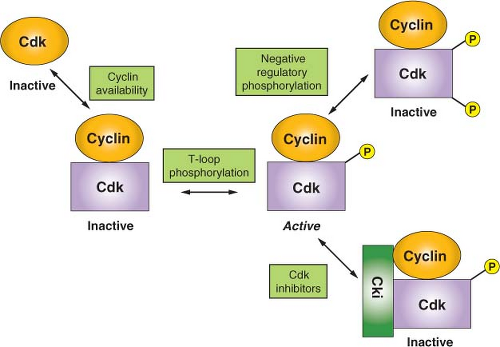 FIGURE 6.2 Principles of cyclin-dependent kinase (CDK) regulation. Because CDK catalytic subunits are inactive when unbound to cyclins, the first level of regulation is through the expression and availability of cyclins. The second level of regulation appears to constitute a housekeeping function in that cyclin binding stimulates an essential phosphorylation of a threonine within a structural feature known as the T loop. Binary complexes with phosphorylated T loops are active. Such active kinase complexes can be subjected to negative regulation in two ways. Additional phosphorylation events on threonine 14 and tyrosine 15 (or the equivalent residues) render the kinase inactive. In addition, the formation of ternary complexes with CDK inhibitory proteins (Ckis) promotes an inactive state. One class of Cki, INK4, specific for CDK4 and CDK6, can inhibit by forming binary complexes directly with the CDK. |
A second important mode of CDK regulation is by phosphorylation. CDKs require an activating phosphorylation on a structural feature designated the T loop. Phosphorylation induces a movement of the T loop that has global effects on CDK structure, including an increase in CDK-cyclin contacts and changes in the substrate-binding site.18 In most if not all instances, T-loop phosphorylation appears to constitute a housekeeping function that occurs concomitant with cyclin binding. However, negative regulatory phosphorylation of CDKs is a highly dynamic process. Proper cell-cycle regulation of CDK1, in particular, requires phosphorylation on two residues within the N-lobe, adjacent to the ATP-binding site: threonine 14 and tyrosine 15. During the normal course of the cell cycle, as cyclin B-CDK1 complexes accumulate, they are immediately phosphorylated at these sites and thereby kept inactive. This allows stockpiling of the large numbers of cyclin B-CDK1 complexes required for efficient entry into mitosis and maintaining them in an inactive state during late S phase and G2. At the G2/M-phase boundary, there is concerted dephosphorylation of these residues, causing cells to advance rapidly into mitosis. Although CDK2 and CDK4 have also been reported to undergo negative regulatory phosphorylation at the homologous residues, the function(s) of this regulation are not as clear-cut (but see “DNA Damage Checkpoints”).
A third mode of CDK regulation is through the action of inhibitory proteins that can form either binary complexes with CDKs or ternary complexes with cyclin-CDK dimers. These exist in three major families. The INK4 family consists of four members (p15, p16, p18, and p19). All are composed of a series of conserved structural motifs known as ankyrin repeats and they specifically target CDK4 and CDK6. The mechanism of action of these inhibitors is to bind the CDK subunit and, by causing a rotation of the N-lobe relative to the C-lobe, constraining the kinase in an
inactive conformation and, in addition, precluding cyclin binding.19 The Cip/Kip family consists of three members in mammals: p21Cip1, p27Kip1, and p57Kip2. All contain a conserved amino terminal cyclin-CDK-binding inhibitory domain and a divergent C-terminal domain possessing other less well-characterized functions. Although these have been characterized primarily as potent inhibitors of CDK2 and have more recently been shown to also be effective CDK1 inhibitors, the case for inhibition of CDK4 and CDK6 is less certain. Whereas Cip/Kip inhibitors are clearly capable of inhibiting CDK4 and CDK6 at high concentration, it is not clear that these conditions are met in vivo, and the situation is further complicated by the finding that Cip/Kip inhibitor binding is actually required to provide a chaperonin or assembly function for the efficient formation of active cyclin D-CDK4 complexes.20 In the case of cyclin A-CDK2, where structural studies have been carried out, it appears that the Cip/Kip inhibitors first anchor via a high-affinity interaction with the cyclin.21 This then allows the inhibitor polypeptide to invade and deform the N-lobe, thus interfering with ATP binding and catalysis.16 The final class of inhibitors consists of two members of the pRb protein family, p107 and p130. Although these proteins have well-characterized functions as transcriptional inhibitors, they also are potent cyclin E/A-CDK2 inhibitors. p107 and p130 each contain cyclin-binding and CDK-binding sites that collaborate to confer inhibitory activity.
inactive conformation and, in addition, precluding cyclin binding.19 The Cip/Kip family consists of three members in mammals: p21Cip1, p27Kip1, and p57Kip2. All contain a conserved amino terminal cyclin-CDK-binding inhibitory domain and a divergent C-terminal domain possessing other less well-characterized functions. Although these have been characterized primarily as potent inhibitors of CDK2 and have more recently been shown to also be effective CDK1 inhibitors, the case for inhibition of CDK4 and CDK6 is less certain. Whereas Cip/Kip inhibitors are clearly capable of inhibiting CDK4 and CDK6 at high concentration, it is not clear that these conditions are met in vivo, and the situation is further complicated by the finding that Cip/Kip inhibitor binding is actually required to provide a chaperonin or assembly function for the efficient formation of active cyclin D-CDK4 complexes.20 In the case of cyclin A-CDK2, where structural studies have been carried out, it appears that the Cip/Kip inhibitors first anchor via a high-affinity interaction with the cyclin.21 This then allows the inhibitor polypeptide to invade and deform the N-lobe, thus interfering with ATP binding and catalysis.16 The final class of inhibitors consists of two members of the pRb protein family, p107 and p130. Although these proteins have well-characterized functions as transcriptional inhibitors, they also are potent cyclin E/A-CDK2 inhibitors. p107 and p130 each contain cyclin-binding and CDK-binding sites that collaborate to confer inhibitory activity.
A final mode of CDK regulation is via control of nuclear import/export. This level of regulation is most obvious for cyclin B-CDK1 complexes, which are kept out of the nucleus via active nuclear export until late G2, when phosphorylation inactivates cis-acting nuclear export signals allowing nuclear accumulation.22 Sequestration of cyclin B-CDK1 in the cytoplasm is a redundant mechanism, along with negative regulatory phosphorylation of CDK1, for preventing premature phosphorylation of mitotic targets.
INDUCTION OF CELLCYCLE PHASE TRANSITIONS
The cell cycle is composed of two action phases, S phase and M phase, in which the genetic material is duplicated and the components of a mother cell are divided into two daughter cells, respectively. The intervening phases, G1 and G2, are thought to exist primarily to allow time for cell growth and for regulatory inputs. Therefore, from the point of view of regulatory theory, cell proliferation is controlled operationally at two key transitions: that between G1 and S phase and that between G2 and M phase. The important characteristic of these two transitions is that, once initiated based on integration of regulatory signals, they must be executed decisively to maintain genetic and genomic integrity. This is accomplished by using a combination of positive and negative modulators to set up the equivalent of a molecular capacitor.
In cycling mammalian cells, the programmed accumulation of cyclins E and A via transcriptional induction provides the positive impetus for the G1-S phase transition. However, these kinases are kept in check by the action of Cip/Kip family inhibitors. If the internal and external environments are permissive for proliferation, the continued accumulation of cyclins will eventually titrate the inhibitors, allowing the latter to be phosphorylated by free cyclin-CDK complexes. Phosphorylation then marks these inhibitors as targets of ubiquitinmediated proteolysis. The concerted destruction of CDK inhibitors and concomitant activation of the entire pool of CDK complexes assure that the transition into S phase is rapid and irreversible.
Although the details of its regulation are somewhat different, the strategy underlying control of the G2-M transition is similar. Cyclin B-CDK1 complexes accumulate starting near the end of S phase but are held in check not by CDK inhibitors but by negative regulatory phosphorylation of CDK1. This phosphorylation on threonine 14 and tyrosine 15 is carried out by kinases Wee1 and Myt1. Entry into M phase is signaled by the rapid dephosphorylation of T14 and Y15, resulting in activation of CDK1. This dephosphorylation is carried out by specialized protein phosphatases, CDC25B and CDC25C. The concerted dephosphorylation of CDK1 depends on activation of CDC25 isoforms by phosphorylation, as well as ubiquitin-mediated proteolysis of Wee1, also in response to phosphorylation. Although the initial activation of CDC25 isoforms is thought to be carried out by other protein kinases, such as Plk1, CDC25B and C are also activated by cyclin B-CDK1, establishing a positive feedback loop. These positive feedback dynamics leading to the simultaneous activation of a large accumulated pool of cyclin B-CDK1 assures that entry into mitosis is decisive. The turnover of Wee1 enforces irreversibility. Because entry into mitosis involves dismantling many of the cell’s components and organelles, as well as construction and use of a complex apparatus for segregating the cell’s genetic material, mitosis is a period of particular vulnerability, and therefore it is important that this transition and subsequent events be carried out rapidly and efficiently.
An important secondary transition that occurs within M phase is that between metaphase and anaphase (Fig. 6.3).23 To preserve genomic integrity, all duplicated chromosomes must be aligned along the cell’s equator and properly attached to microtubules of the mitotic spindle. The trigger for separation of sister chromatids and their movement to opposite poles of the cell is the activation of the protein-ubiquitin ligase APC/C. This
is achieved via CDK1 activation, but more importantly by the binding of a key cofactor, CDC20, whose availability is linked to the proper attachment of chromosomes and the integrity of the spindle. The targets of the APC/C are a protein known as securin as well as cyclin B, both of which inhibit a specialized protease, separase. The key target of separase is a complex that binds sister chromatids together: cohesin. Cleavage of the Scc1 subunit of cohesin leads to a rapid execution of anaphase. It is the ability to stockpile a large pool of inactive securin-separase complexes that can be rapidly mobilized by irreversible proteolysis of securin and cyclin B that allows for a rapid irreversible metaphase-anaphase transition.
is achieved via CDK1 activation, but more importantly by the binding of a key cofactor, CDC20, whose availability is linked to the proper attachment of chromosomes and the integrity of the spindle. The targets of the APC/C are a protein known as securin as well as cyclin B, both of which inhibit a specialized protease, separase. The key target of separase is a complex that binds sister chromatids together: cohesin. Cleavage of the Scc1 subunit of cohesin leads to a rapid execution of anaphase. It is the ability to stockpile a large pool of inactive securin-separase complexes that can be rapidly mobilized by irreversible proteolysis of securin and cyclin B that allows for a rapid irreversible metaphase-anaphase transition.
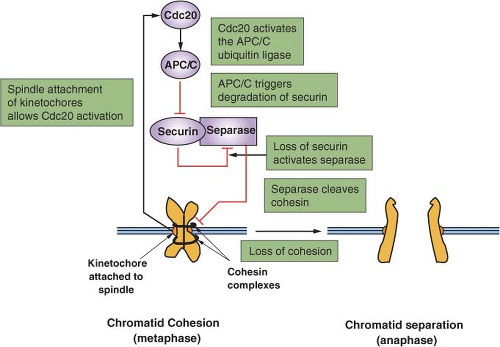 FIGURE 6.3 Regulation of the metaphase-anaphase transition. After replication, paired sister chromatids are held together by a complex of proteins, known collectively as cohesin, thus preventing anaphase. However, once paired chromatids have formed bivalent attachments to a functional mitotic spindle, inhibition of CDC20 mediated by the chromosome kinetochores is relieved. CDC20 is an essential cofactor of the protein-ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C), which now becomes active. The primary initial target of the APC/C is securin, an inhibitor of the protease separase. Ubiquitinmediated proteolysis of securin releases separase to cleave the cohesin subunit Scc1. This event releases paired chromatids from cohesion, thereby allowing anaphase to proceed. |
UBIQUITIN-MEDIATED PROTEOLYSIS
It is becoming increasingly evident that much of the regulation of cell-cycle phase transitions depends on ubiquitin-mediated proteolysis.24 Ubiquitin is a 76-amino acid polypeptide that can be covalently linked to lysines of other proteins via the formation of an isopeptide bond with its C-terminal carboxylate. Additional ubiquitin molecules can then be attached to the lysines of already conjugated ubiquitin to form polyubiquitin chains. Polyubiquitylated proteins are usually targeted for rapid proteolysis by a large multisubunit protease known as the proteasome. The enzymes that transfer ubiquitin to target proteins are known as protein-ubiquitin ligases. From the perspective of cellcycle control, two families of protein-ubiquitin ligases have predominant roles. The first family, SCF (Skp1-Cullin-F-box protein), specifically targets proteins that are marked for destruction by phosphorylation. This allows degradation of specific proteins to be regulated at a separate level from the protein-ubiquitin ligase itself, which can be expressed constitutively and used to target a large number of substrates independently. SCF ligases consist of three invariant core components and one of a number of specificity factors (F-box proteins) that recognize phosphorylated substrates. A few notable examples of SCF proteinubiquitin ligases are SCFSkp2 (containing the F-box protein Skp2), which targets p2725 and p130,26
and SCFCDC4 (containing the F-box protein CDC4, also known as FBXW7), which targets cyclin E.27,28,29 The second family of protein-ubiquitin ligases that is critical for cell-cycle control is known collectively as the APC/C. The APC/C is a large complex consisting of 12 core subunits and 1 of 2 specificity factors, CDC20 and CDH1. Unlike SCF ubiquitin ligases, targeting of substrates by the APC/C is determined by ligase activation rather than substrate activation. APCCDC20 is active from metaphase until the end of mitosis as a result of periodic accumulation and degradation of CDC20 itself. APCCDH1, on the other hand, which is negatively regulated by CDK-mediated phosphorylation, is activated on mitotic exit and during the subsequent G1 interval when CDKs are inactive. In this manner, important mitotic targets, such as cyclin A, cyclin B, and securin (as well as many others), are degraded during mitosis and prevented from reaccumulating during the subsequent G1 interval.
and SCFCDC4 (containing the F-box protein CDC4, also known as FBXW7), which targets cyclin E.27,28,29 The second family of protein-ubiquitin ligases that is critical for cell-cycle control is known collectively as the APC/C. The APC/C is a large complex consisting of 12 core subunits and 1 of 2 specificity factors, CDC20 and CDH1. Unlike SCF ubiquitin ligases, targeting of substrates by the APC/C is determined by ligase activation rather than substrate activation. APCCDC20 is active from metaphase until the end of mitosis as a result of periodic accumulation and degradation of CDC20 itself. APCCDH1, on the other hand, which is negatively regulated by CDK-mediated phosphorylation, is activated on mitotic exit and during the subsequent G1 interval when CDKs are inactive. In this manner, important mitotic targets, such as cyclin A, cyclin B, and securin (as well as many others), are degraded during mitosis and prevented from reaccumulating during the subsequent G1 interval.
REGULATION OF THE CELL CYCLE
To preserve organismic function and integrity, the cell cycle must be regulated at a number of levels. These include entry into and exit from proliferation mode, coordination of cell-cycle events, and specialized responses that increase the probability of surviving a variety of environmental and internally generated insults.
Quiescence and Differentiation
The most fundamental aspect of cell-cycle control is the regulation of entry and exit. For mammalian cells, the decision to enter or exit the proliferative mode is based on environmental signals such as mitogens, growth factors, hormones, and cell-cell contact, as well as on internal differentiation programs. If the state of cell-cycle exit is reversible, it is referred to as quiescence. If it is in the context of terminal differentiation, cell-cycle exit may merely be one component of a differentiation program. Although cells entering quiescence and postmitotic differentiation vary from each other in many respects, from the perspective of cell-cycle control, they have much in common. First, cell-cycle exit is usually associated, at least initially, with an accumulation of G1/S CDK inhibitors. Members of the INK4 family, targeting CDK4 and CDK6 and members of the Cip/Kip family, as well as the Rb-related protein p130, all targeting CDK2, are up-regulated. This causes accumulation of cells in G1, from where cell-cycle exit can occur. Next, or simultaneously, the positive cell-cycle machinery is dismantled by downregulation of CDKs and cyclins, primarily at the transcriptional level. In the case of quiescence, cell-cycle exit is paralleled by a reduced rate of protein synthesis, indicative that cells have entered a resting state.
Entry into and exit from quiescence are mediated largely by growth factors and mitogens that interact with cell surface receptors. These in turn are linked to intracellular signaling cascades that up-regulate the rate of protein synthesis as well as the transcription of genes that promote proliferation, such as those encoding CDKs and cyclins. The two best-characterized signaling pathways in this context are the mitogen-activated protein kinase/extracellular signaling-regulated kinase pathway30 and the phosphoinositide 3 (PI3) kinase/ AKT pathway,31 shown in Figure 6.4




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
