Cell Biology of Insulin Secretion
Jean-Claude Henquin
Under physiologic conditions, the concentration of blood glucose fluctuates only in a narrow range despite alternations in periods of food intake and fasting. This stability is due to a remarkably efficient hormonal system that exerts opposite effects on the organs of glucose storage and production. Whereas several hormones can prevent dangerous declines in blood glucose concentrations by stimulating glycogenolysis and gluconeogenesis, insulin secretion by β-cells of the islets of Langerhans is the only efficient means by which the organism can decrease the blood glucose concentration. Any alteration in the β-cell functioning has thus a profound impact on glucose homeostasis: excessive secretion of insulin causes hypoglycemia, and insufficient secretion leads to diabetes mellitus. It is therefore not surprising that insulin secretion is subject to tight control. This control is ensured by glucose itself and by an array of metabolic, neural, hormonal, and sometimes pharmacologic factors, which necessitates rapid integration by β-cells of a host of signals generating intracellular messengers.
Stimulus-secretion coupling in β-cells is astonishingly complex and still incompletely understood despite continuous investigation. However, its main features are beginning to appear clearly enough to gain wide acceptance. Only these major characteristics will be discussed here, leaving issues that are too controversial and specialized for more specific review articles. Because this chapter is devoted to normal stimulus-secretion coupling, I have given preference to publications of studies performed with normal β-cell models and limited those of studies using insulin-secreting cell lines that often show abnormal properties. Although in vitro studies of normal adult human islets are still few, they indicate that the information gained from animal experiments can be extrapolated in large part to humans. Only functionally important specificities of the human β-cell will thus be highlighted.
IDENTIFICATION OF GLUCOSE BY β-CELLS
In most secretory cells, extracellular stimuli bind to membrane receptors and activate stereotyped transduction pathways that eventually trigger specific responses. The existence in the plasma membrane of β-cells of a glucoreceptor that would trigger insulin secretion upon binding of glucose itself has been considered but could never be proved. On the other hand, compelling evidence indicates that glucose must be metabolized by β-cells to induce insulin secretion. Small interferences with glucose metabolism have repercussions on insulin secretion, whereas marked interferences with insulin secretion may be without effect on glucose metabolism in β-cells. The experimental work on which this “fuel-concept” of insulin secretion is based has been discussed in detail in several review articles (12345678).
β-Cells detect blood glucose variations and adjust the rate of insulin secretion to these variations through changes in their metabolism. This ability rests on a peculiar biochemical organization that is summarized in Figure 6.1.
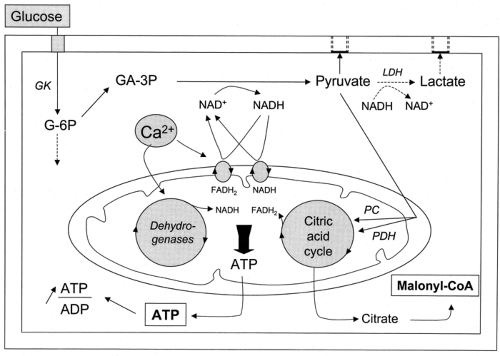 Figure 6.1. Schematic summary of glucose metabolism in the cytoplasm and mitochondria of β-cells. GK, glucokinase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; PC, pyruvate carboxylase; G-6P, glucose-6-phosphate; GA-3P, glyceraldehyde-3-phosphate; NAD+ and NADH, oxidized and reduced nicotinamide adenine dinucleotide, respectively; FADH2, reduced flavin adenine dinucleotide; ATP, adenosine triphosphate; ADP, adenosine diphosphate; CoA, coenzyme A. |
In rodent β-cells, as in hepatocytes, glucose entry is mediated by the high-capacity, low-affinity (Km ∼17 mM) glucose
transporter GLUT2 (9,10). In human β-cells, GLUT2 is less extensively expressed, and GLUT1 predominates (11,12). Nevertheless, whatever the proportion of the different transporters, glucose transport is always efficient enough to permit rapid equilibration between the intracellular and extracellular concentrations of glucose (13,14). The rate of glucose transport exceeds the rate of glucose utilization and is, therefore, not limiting (10,15,16).
Phosphorylation of glucose into glucose-6-phosphate is catalyzed by a glucokinase (GK hexokinase IV) similar (15 amino acids shorter) to that present in hepatocytes (17,18). The same gene is controlled by two distinct promoters, permitting differential regulation of the expression—positively by insulin and negatively by glucagon in the liver and positively by glucose in β-cells. The characteristics of GK, a Km between 6 and 10 mM, sigmoidal dependency on its substrate concentration, and lack of inhibition by its product (glucose-6-phosphate), make it a suitable glucose sensor (15). The view that GK is the flux-determining step of glucose metabolism in β-cells is supported by the fact that its activity is lower than that of the other potentially regulatory enzymes of glycolysis and closely matches glucose usage (10,15,16). Mutations of the GK gene result in the production of either hypoactive isoforms of the enzyme in patients with maturity-onset diabetes of the young (MODY-2) (19) or hyperactive isoforms in glucokinase-linked hyperinsulinemia and hypoglycemia (20). Expression of these mutants in insulin-secreting cell lines produced the expected decreasing and increasing effects on glucose metabolism and insulin secretion (21). Whereas the important role of GK is admitted unanimously, the concept that the enzyme is the sole regulator of glycolysis in β-cells remains disputed (22). The contribution of a high-affinity hexokinase has often been considered, but recent experiments have cast doubt on the presence of this enzyme in β-cells (23).
Under physiologic conditions, dephosphorylation of glucose-6-phosphate by glucose-6-phosphatase, storage of glucose as glycogen, and transformation of glucose to sorbitol are quantitatively small (4,5,16,22,24). They play no significant role in the control of normal β-cell function. Glucose metabolism through the pentose phosphate pathway does not exceed 5%, and its relative contribution decreases as the glucose concentration is increased. This pathway also is thought to be unimportant for the control of insulin secretion. Glycolysis is the major route of glucose metabolism in β-cells.
β-Cells contain the muscle (PFK1-M) and cerebral (PFK1-C) isoforms of phosphofructokinase (25). The characteristics of the enzyme and of its regulators indicate that PFK adjusts its rate of phosphorylation to that of glucokinase (15,16). Normal β-cells hardly express lactate dehydrogenase (24,26), and plasma membrane monocarboxylate transporters (for lactate and pyruvate) are present in only low amounts (26,27), so that pyruvate generated by glycolysis is channeled (>90%) to the mitochondria. However, the low activity of lactate dehydrogenase is insufficient to regenerate enough oxidized nicotinamide adenine dinucleotide (NAD+) to sustain a high glycolytic flux. Reoxidation of NADH is thus achieved by two mitochondrial electron shuttles that are unusually active in β-cells: the glycerol-3-phosphate shuttle and the malate-aspartate shuttle (7,26,28,29). The two shuttles appear to be at least partially redundant but, together, play an essential role in β-cell function (30,31).
Within mitochondria, pyruvate is transformed approximately equally into acetyl coenzyme A (CoA) by pyruvate dehydrogenase and into oxaloacetate by pyruvate carboxylase (32,33). Acetyl CoA is then used by the citric acid cycle, with formation of NADH and reduced flavin adenine dinucleotide (FADH2), which together with NADH and FADH2 issued from the activity of the shuttles previously described, transfer electrons to the electron-transport chain. This eventually results in hyperpolarization of the mitochondrial membrane and generation of adenosine triphosphate (ATP) (34). The high activity of pyruvate carboxylase in β-cells is unusual for a non-gluconeogenic, non-lipogenic tissue. The enzyme serves to fill the citric acid cycle with intermediates (anaplerosis) (35), which allows some of these intermediates to exit from the mitochondria without compromising the cycle function. These compounds can then be used as precursors of amino acids (24) or, in the case of citrate, produce putative second messengers such as malonyl CoA (36) (see below).
When the concentration of glucose is raised, the acceleration of glucose oxidation is greater than that of glycolysis (22). This remarkable feature is attributed to the activation of mitochondrial dehydrogenases. Three of these, pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase, are activated by the increase in mitochondrial Ca2+ that follows the increase in cytoplasmic Ca2+ induced by glucose (37) (see below). A fourth dehydrogenase, the FAD-linked mitochondrial glycerol 3-P-dehydrogenase, the key enzyme of the glycerol phosphate shuttle (see point 4 above), is also Ca2+-sensitive, but its activation is achieved by the increase in cytoplasmic Ca2+.
The major consequence of this original biochemical organization of β-cells is that the ATP/adenosine diphosphate (ADP) ratio increases with the glucose concentration (38). It is important to emphasize how this situation is exceptional. Thus, most cells, including glucagon-secreting α-cells, adapt their metabolism to maintain a constant ATP/ADP ratio when the concentration of glucose changes (39). The glucose-induced variations in ATP/ADP ratio observed in purified β-cells or in whole islets take place in the cytoplasm. They are often underestimated because of a partial masking by a stable pool of nucleotides within insulin granules, occur over a wide range of glucose concentrations and are rapid, preceding any increase in cytoplasmic [Ca2+]i(38,40,41). All these characteristics support the proposal that adenine nucleotides serve as second messenger in stimulus-secretion coupling.
This metabolic recognition of glucose by β-cells is then coupled to the process of insulin secretion through the operation of two pathways: a triggering pathway and an amplifying pathway (42). Their major steps are schematized in Figure 6.2.
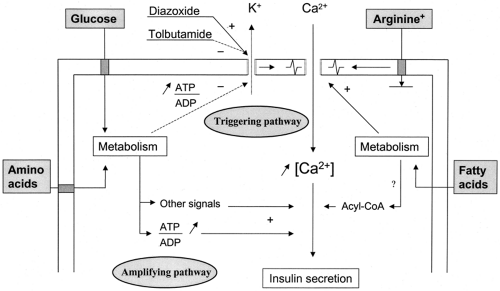 Figure 6.2. Schematic representation of the triggering and amplifying pathways of the stimulation of insulin secretion by glucose, and of the mode of action of other nutrients: +, stimulation; – inhibition; ATP, adenosine triphosphate; ADP, adenosine diphosphate; CoA, coenzyme A. |
THE TRIGGERING PATHWAY OF GLUCOSE-INDUCED INSULIN SECRETION
Cytoplasmic Ca2+ as Triggering Signal
Direct evidence that cytoplasmic Ca2+ triggers exocytosis of insulin granules was obtained from experiments using β-cells of which the plasma membrane was permeabilized. In these cells, the membrane potential is dissipated and the cytosolic concentration of small molecules (e.g., Ca2+, ATP) can be controlled. An increase in free [Ca2+]i induced insulin release independently from any other change (43,44). More recently, exocytosis of insulin granules was estimated by measuring the capacitance of voltage-clamped single β-cells: increases in capacitance reflect increases in plasma membrane surface caused by the fusion of secretory granules. This technique confirmed that Ca2+ directly triggers exocytosis (45,46). Although progressive desensitization
to Ca2+ occurs in these preparations, imposed oscillations of [Ca2+]i were able to entrain oscillations of insulin secretion in permeabilized cells (47), indicating that Ca2+ has a minute-to-minute triggering role.
to Ca2+ occurs in these preparations, imposed oscillations of [Ca2+]i were able to entrain oscillations of insulin secretion in permeabilized cells (47), indicating that Ca2+ has a minute-to-minute triggering role.
Experiments using permeabilized insulin-secreting cells (47) and capacitance measurements in β-cells (48) have shown that stable derivatives of guanosine triphosphate (GTP) can increase insulin release in a truly Ca2+-independent manner. In intact rat islets, a Ca2+-independent pathway of glucose-induced insulin secretion could be unmasked by strong and combined activation of protein kinases A and C (49). However, the significance of this pathway is unclear (50). Under physiologic conditions, glucose increases the cytoplasmic free Ca2+ concentration ([Ca2+]i) in β-cells, and all maneuvers interfering with this increase impair the stimulation of insulin secretion. Although the sequence of events leading to insulin secretion may include Ca2+-independent steps, the physiologic regulation by glucose is achieved through a Ca2+-dependent pathway.
How can cytoplasmic [Ca2+]i serve as triggering signal? Unstimulated β-cells maintain [Ca2+]i between 50 and 100 nM by several mechanisms: a low permeability of the plasma membrane to Ca2+, pumping of Ca2+ into the endoplasmic reticulum by a sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) (51,52), and extrusion of Ca2+ across the plasma membrane by a calmodulin-sensitive Ca2+-ATPase (53,54) and an Na+/Ca2+ countertransport driven by the electrochemical gradient for Na+ (55). There thus exist large gradients of Ca2+ concentration between the cytoplasm and either the lumen of the endoplasmic reticulum or the extracellular medium. Opening of Ca2+ channels in the plasma membrane (e.g., by depolarization) or in the endoplasmic reticulum membranes (e.g., by inositol trisphosphate) allows rapid movements of Ca2+ down the electrochemical gradient and causes increases in cytoplasmic [Ca2+]i.
How Does Glucose Increase [Ca2+]i in β-Cells?
Mobilization of Ca2+ from intracellular stores is the major mechanism by which many stimuli increase [Ca2+]i in electrically non-excitable cells. β-Cells are electrically excitable, and mobilization of intracellular Ca2+ by glucose has never been demonstrated by direct measurements in normal β-cells. Although Ca2+ handling by intracellular organelles contributes to the fine regulation of [Ca2+]i during glucose stimulation (56,57), Ca2+ influx through voltage-operated Ca2+ channels is indisputably the major determinant of the glucose-induced [Ca2+]i increase (Fig. 6.2).
When the extracellular concentration of glucose increases, β-cell metabolism accelerates (Fig. 6.1). One of the earliest consequences is a decrease in the K+ conductance of the plasma membrane (58,59,60). It is due to the closure of the K+ channels that determine the resting potential of β-cells, the ATP-sensitive K+channels (K+-ATP channels) (61,62). These channels are tetramers of a complex of two proteins: a high-affinity sulfonylurea receptor (SUR 1) and an inwardly rectifying K+ channel (Kir 6.2) (63,64,65). The pore of the channel is formed by the four subunits of Kir 6.2 to which ATP binds to close the channel. The four subunits of SUR 1 mediate the opening property of Mg2+-ADP and endow the channel with sensitivity to pharmacologic agents such as sulfonylureas and diazoxide that respectively close and open the channel (Fig. 6.2).
The decrease in K+ conductance produced by glucose allows a background current of a yet unidentified nature to move the membrane potential away from the equilibrium potential for K+, that is, to depolarize the membrane (Fig. 6.3). In a mouse β-cell, within an intact islet, the resting potential is about -65 mV in the presence of a non-stimulatory glucose concentration (3 mM). When the concentration of glucose is increased to 10 mM,
the membrane depolarizes by about 15 mV, to reach the threshold potential for opening of voltage-operated Ca2+ channels (66). Depending on the species, these voltage-operated Ca2+ channels are predominantly or exclusively of the L-type (67,68). Their opening allows the influx of Ca2+, which manifests itself as a rapid depolarization to a plateau potential on which a burst of Ca2+ action potentials appears. The electrical activity then suddenly stops and the membrane repolarizes. The membrane potential then starts to oscillate, thus producing rhythmic openings of Ca2+ channels and Ca2+ influx. As the glucose concentration increases, the phases of depolarization and Ca2+ influx become longer and the intervals become shorter (Fig. 6.3). The membrane eventually remains persistently depolarized at the plateau potential and exhibits continuous electrical activity. This characteristic pattern has been studied primarily in vitro (66) but is also observed when the membrane potential of β-cells is recorded in vivo (69).
the membrane depolarizes by about 15 mV, to reach the threshold potential for opening of voltage-operated Ca2+ channels (66). Depending on the species, these voltage-operated Ca2+ channels are predominantly or exclusively of the L-type (67,68). Their opening allows the influx of Ca2+, which manifests itself as a rapid depolarization to a plateau potential on which a burst of Ca2+ action potentials appears. The electrical activity then suddenly stops and the membrane repolarizes. The membrane potential then starts to oscillate, thus producing rhythmic openings of Ca2+ channels and Ca2+ influx. As the glucose concentration increases, the phases of depolarization and Ca2+ influx become longer and the intervals become shorter (Fig. 6.3). The membrane eventually remains persistently depolarized at the plateau potential and exhibits continuous electrical activity. This characteristic pattern has been studied primarily in vitro (66) but is also observed when the membrane potential of β-cells is recorded in vivo (69).
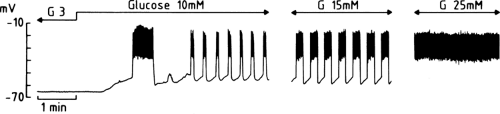 Figure 6.3. Changes in the membrane potential of a mouse β-cell induced by successive increases in the concentration of glucose in the perifusion medium. The three recordings were obtained in the same cell. (Adapted from Henquin JC. Les mécanismes de contrôle de la sécrétion d’insuline. Arch Int Physiol Biochim 1990;98:A61-A80, with permission.) |
Attributing the electrical properties of β-cells only to K+-ATP channels and voltage-operated Ca2+ channels is an oversimplification imposed by the objectives of this chapter. The electrogenic sodium pump and a number of voltage-dependent and voltage-independent ionic channels also participate in the fine tuning of the glucose-induced electrical activity, which is still only partly understood (66,68,70,71). Whether the changes in the ATP/ADP ratio suffice to explain the influence of glucose on the β-cell membrane potential is also uncertain. The fact that the ratio gradually changes between 0 and 20 mM glucose (38) makes the hypothesis at least plausible. Functionally, the decrease in ADP (which acts on SUR 1) is probably more important than the increase in ATP. Other metabolic regulators of the membrane potential have been proposed, but none of these suggestions has gained wide acceptance (63,64,65).
Characteristics of the Increase in [Ca2+]i Produced by Glucose
In the presence of a non-stimulatory concentration of glucose, β-cell [Ca2+]i is low and stable. Upon stimulation with high glucose, [Ca2+]i changes in three stages (Fig. 6.4A). A small decrease occurs initially, reflecting cytoplasmic Ca2+ pumping into the endoplasmic reticulum (72). This Ca2+ sequestration might be promoted by the increase in ATP/ADP ratio (73), but its functional significance is unknown. The subsequent [Ca2+]i increase displays a biphasic pattern. An initial large and sustained increase is followed by a nadir and then by oscillations that persist as long as the glucose stimulation is continued. These changes in [Ca2+]i are superimposable on those of the membrane potential (compare Fig. 6.4A and Fig. 6.3) (74,75). The initial decrease in [Ca2+]i occurs during progressive depolarization from the resting to the threshold potential, but the two events do not seem to be causally related (76). In contrast, the long first phase of the [Ca2+]i increase and the subsequent oscillations not only coincide with but are caused by the influx of Ca2+ brought about by the changes in membrane potential. The mechanisms of the oscillations in β-cell membrane potential (66,68,70,71) and [Ca2+]i (77,78,79) during steady-state stimulation with glucose remain controversial. They seem to result from a complex interplay between oscillations in metabolic signals and the coordinated opening and closing of ionic channels with distinct time- and voltage-dependent properties. Ca2+-independent, intrinsic oscillations of glucose metabolism (77), Ca2+-induced acceleration of ATP production (80), and Ca2+-induced stimulation of ATP consumption (40) have been proposed. The variability of these oscillations (period from 20 seconds to 4 minutes) between preparations adds to the complexity. Sometimes, glucose-induced electrical activity and [Ca2+]i oscillations are characterized by a double periodicity in which rapid oscillations are superimposed on slower ones (Fig. 6.4B). It is not unusual that, in the same islet, the oscillatory pattern spontaneously changes from regular, fast oscillations into mixed or slow oscillations. Cyclic adenosine monophosphate (cAMP) increases the rhythm and regularity of the oscillations.
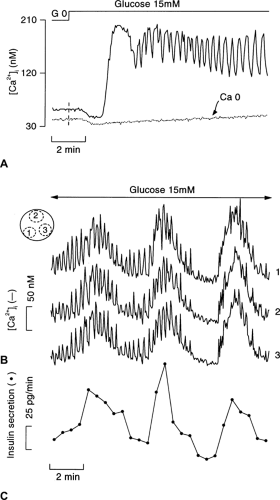 Figure 6.4. Glucose-induced changes in cytoplasmic free Ca2+ ([Ca2+]i) in intact mouse islets. A: After an initial period of perifusion without glucose (G 0), the islet was stimulated with 15 mM glucose. The medium contained 2.5 mM or 0 mM CaCl2. B: [Ca2+]i and insulin secretion were measured simultaneously in the same mouse islet during continuous stimulation with 15 mM glucose. Traces 1–3 correspond to the [Ca2+]i signal recorded in the different regions of the islet schematically identified in the upper left corner. They show the excellent synchrony of both slow and rapid oscillations of [Ca2+]i. The lower line shows that insulin secretion (sampling every 30 seconds) oscillates in phase with the slow [Ca2+]i oscillations. A higher sampling rate would be necessary to show that the fast [Ca2+]i oscillations also trigger oscillations of insulin secretion (experiments performed by M. A. Ravier). |
Figure 6.4 A also shows that glucose does not increase [Ca2+]i in the absence of extracellular Ca2+. This is also true when, in the presence of extracellular Ca2+, Ca2+ influx is impeded by blocking the voltage-operated Ca2+ channels or by preventing the glucose-induced depolarization with diazoxide (see below).
Activation of an Effector System by Ca2+
Insulin is stored in large dense core vesicles. Its secretion in the extracellular space of the islets occurs by a process of regulated exocytosis (81) that involves several steps: recruitment of the granules, followed by their docking, priming, and eventual fusion with the plasma membrane. Extensive description of these complex stages can be found in specialized review articles (82,83).
The importance of the cytoskeleton has been established by the use of drugs capable of interfering with the function of its distinct elements (84,85). The cytoskeleton, composed of microfilaments, microtubules, and intermediary filaments (vimentin, cytokeratin), ensures the transport of the secretory granules to the plasma membrane. Upon β-cell stimulation, tubulin polymerizes to form microtubules that can guide the granules toward the sites of exocytosis. Polymerization of globular actin with formation of microfilaments also occurs. Actin filaments may both facilitate and impede (cortical network) the access of
secretory granules to the plasma membrane. Ca2+-calmodulin protein kinase II (Cam-kinase II), associated with the secretory granules, appears to play an important role in this mobilization of the granules from the Golgi to the exocytotic sites (86). The activation of Cam-kinase II by the increase in [Ca2+]i that glucose and other secretagogues produce correlates well with insulin secretion. Myosin light-chain kinase also is implicated in granule movements by modulation of actin-myosin interactions (87).
secretory granules to the plasma membrane. Ca2+-calmodulin protein kinase II (Cam-kinase II), associated with the secretory granules, appears to play an important role in this mobilization of the granules from the Golgi to the exocytotic sites (86). The activation of Cam-kinase II by the increase in [Ca2+]i that glucose and other secretagogues produce correlates well with insulin secretion. Myosin light-chain kinase also is implicated in granule movements by modulation of actin-myosin interactions (87).
The docking step corresponds to the targeting of the secretory vesicle to the plasma membrane (82,83). It is achieved by the pairing of cognate proteins present on the vesicle membrane (v-SNARE, e.g., synaptobrevin) and the plasma (target) membrane (t-SNARE, e.g., syntaxin). This assembly then recruits several cytoplasmic proteins to reorganize in a multimeric complex through an ATP-dependent process (priming). Finally, Ca2+ acts on yet other proteins of the vesicle membrane, among which synaptotagmin is predominant, to induce the fusion of the two membranes. Eventually, the granule content, i.e., insulin, C peptide, other proteins, and nucleotides, is released outside the β-cell.
GTP-binding proteins also control the process of exocytosis (82,83), but it is unclear whether these serve a regulatory role in glucose-induced insulin secretion. As already mentioned,
experiments using permeabilized insulin-secreting cells (47) or β-cells patch-clamped in the whole-cell mode (48) (to control the cytosolic composition) have shown that insulin release can be induced by GTP, independently of [Ca2+]i changes. The G-protein involved in this stimulation often is referred to as GE (for exocytosis), but its nature is unknown. Heterotrimeric G-proteins can influence insulin secretion independently of their role in membrane receptor-activated signal transduction (88). Gαi or Gαo mediate an inhibitory effect that abolishes the action of Ca2+ (89,90). It has been suggested that the protein phosphatase calcineurin is implicated in this inhibition, but the hypothesis has not been confirmed.
experiments using permeabilized insulin-secreting cells (47) or β-cells patch-clamped in the whole-cell mode (48) (to control the cytosolic composition) have shown that insulin release can be induced by GTP, independently of [Ca2+]i changes. The G-protein involved in this stimulation often is referred to as GE (for exocytosis), but its nature is unknown. Heterotrimeric G-proteins can influence insulin secretion independently of their role in membrane receptor-activated signal transduction (88). Gαi or Gαo mediate an inhibitory effect that abolishes the action of Ca2+ (89,90). It has been suggested that the protein phosphatase calcineurin is implicated in this inhibition, but the hypothesis has not been confirmed.
In addition to Cam-kinase II, two other protein kinases have a marked influence on the effector system. The cAMP-dependent protein kinase A and the Ca2+-and phospholipid-dependent protein kinase C strongly amplify the action of Ca2+ on exocytosis. Their targets are not known (see below).
Consensus Model of the Triggering Pathway
The entry of glucose in β-cells by facilitated diffusion being not rate limiting, changes in the extracellular (blood) glucose concentration are immediately transduced into changes in β-cell metabolism. The tight coupling between glycolysis and mitochondrial metabolism results in rapid and marked changes in the ATP/ADP ratio that then control K+-ATP channels in the plasma membrane. These channels serve as transducers of biochemical into biophysical signals. Thus, their closure by an increase in the ATP/ADP ratio leads to membrane depolarization and opening of voltage-operated Ca2+ channels. The ensuing Ca2+ influx raises [Ca2+]i, which eventually activates an effector system responsible for the exocytosis of insulin granules (Fig. 6.2).
THE AMPLIFYING PATHWAY OF GLUCOSE-INDUCED INSULIN SECRETION
In addition to the generation of a triggering signal, an increase in [Ca2+]i, glucose-induced insulin secretion involves the generation of signals that amplify the action of Ca2+ on the exocytotic process (42) (Fig. 6.2). This concept was established by experiments showing that glucose can still increase insulin secretion when K+-ATP channels cannot be closed (91,92,93) or are already completely closed (94,95,96).
Identification of the Pathway
In a control medium, a high glucose concentration increases [Ca2+]i in β-cells (triggering signal) and stimulates insulin secretion. In a low glucose medium, tolbutamide mimics the effects of glucose. When the drug is used at a maximally effective concentration that closes all K+-ATP channels (Fig. 6.2), it increases [Ca2+]i more than does high glucose but has a less potent effect on insulin secretion (Fig. 6.5). Increasing the glucose concentration under these conditions considerably amplifies insulin secretion without affecting [Ca2+]i. However, glucose does not have this effect when the tolbutamide-induced [Ca2+]i increase has been prevented by omission of extracellular Ca2+.
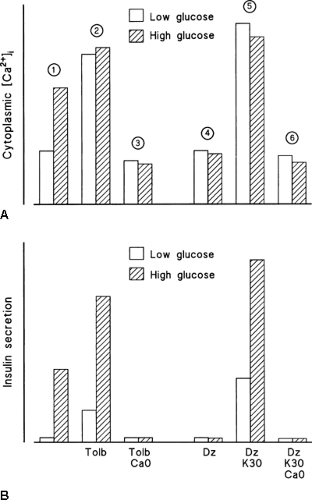 Figure 6.5. Demonstration of the existence of two pathways of control of insulin secretion by glucose. The figure is based on data obtained with mouse islets. Changes in cytoplasmic free Ca2+ ([Ca2+]i) (A) and in insulin secretion (B) under the following conditions are shown: (1) In control medium, high glucose raises cytoplasmic [Ca2+]i and stimulates insulin secretion. (2) Blockade of all K+-ATP channels by a high concentration of tolbutamide (Tolb) in low glucose mimics both effects of high glucose alone, but the stimulation of secretion is smaller in spite of a larger rise in [Ca2+]i. High glucose does not affect [Ca2+]i but markedly increases insulin secretion. (3) The effects of tolbutamide and glucose are abolished when [Ca2+]i cannot be increased because of omission of extracellular Ca2+. (4) Opening of K+-ATP channels by diazoxide (Dz) prevents the stimulatory effects of high glucose on [Ca2+]i and insulin secretion. (5) Depolarization with high K+ strongly increases [Ca2+]i and induces insulin secretion in low glucose. High glucose markedly amplifies the secretory response without changing [Ca2+]i. (6) The effects of high K+ and glucose are abrogated when [Ca2+]i cannot be raised because of omission of extracellular Ca2+. In conclusion, glucose stimulates insulin secretion by increasing [Ca2+]i (triggering pathway as shown in 1), and by increasing the efficacy of Ca2+ on exocytosis (amplifying pathway as shown in 2 and 5). |
When K+-ATP channels are opened by diazoxide (Fig. 6.2), a high glucose concentration is unable to increase [Ca2+]i and stimulate insulin secretion because it cannot depolarize the β-cell membrane (Fig. 6.5). An increase in [Ca2+]i can, however, be induced, even in a low glucose environment, by depolarizing the membrane with high extracellular K+. This is followed by a stimulation of insulin secretion simply because a triggering signal has been produced. Increasing the concentration of glucose in the presence of high K+, while K+-ATP channels are held open by diazoxide, leads to a marked increase in insulin secretion without any further increase in [Ca2+]i (Fig. 6.5). Again, these effects of glucose require that [Ca2+]i be increased, because they are abolished by omission of extracellular Ca2+.
Mechanisms of the Amplifying Pathway
The amplifying pathway requires the metabolism of glucose by β-cells (91). The second messenger has not been identified with certainty, but good correlations exist between the ATP/ADP ratio and glucose-induced insulin secretion through this pathway (38,92). Moreover, several experimental approaches have established that ATP influences insulin secretion at steps distal to the increase in [Ca2+]i (98,99,100). There is also good evidence that cAMP-dependent protein kinase A and diacylglycerol-activated protein kinase C are not involved (92,101,102). Whether long-chain acyl CoAs participate in this effect is still disputed (101,103). More complete discussion of this question can be found elsewhere (42).
Hierarchy between the Triggering and Amplifying Pathways
Under control conditions, the concentration dependency of glucose-induced insulin secretion displays a sigmoidal shape (Fig. 6.6A). A similar relationship characterizes generation of the triggering signals, electrical activity, and increase in [Ca2+]i by glucose (104,105,106). The threshold corresponds to the glucose concentration required to depolarize the β-cell membrane to the potential where voltage-operated Ca2+ channels start to open and the triggering signal thus starts to be produced. In contrast, the concentration dependency of the amplifying pathway, which can be established when the triggering signal is kept constant by high K+ in the presence of diazoxide, is hyperbolic and shifted to the left (Fig. 6.6B). Low concentrations of glucose are thus able to influence insulin secretion, but their influence manifests itself only when a triggering signal has been produced. The similarity of the concentration dependencies of glucose-induced triggering signal and secretory response establishes a clear hierarchy between the two pathways: The amplifying pathway remains functionally silent as long as the triggering pathway has not depolarized the membrane and increased [Ca2+]i, but its role is essential to optimize the secretory response to the triggering signal (42). This hierarchy ensures that no insulin is inappropriately secreted in the presence of low glucose. It may be altered when the function of K+-ATP channels is perturbed by genetic alterations or by long-acting sulfonylureas (42).
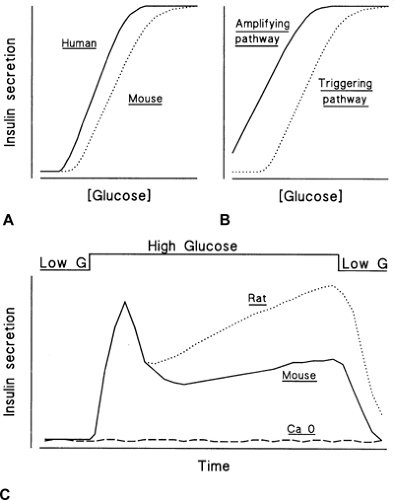 Figure 6.6. Concentration dependence and kinetics of glucose-induced insulin secretion in vitro. A: The relationship between the glucose concentration and insulin secretion is sigmoidal; it is shifted to the left in humans as compared with the curve in mice; the curve would be intermediate in the rat. B: The triggering and amplifying pathways of glucose control of insulin secretion show distinct dependences on the glucose concentration. C: An abrupt increase in the glucose concentration triggers a biphasic secretion of insulin. During second phase, the rate of secretion increases more in the rat than in the mouse. Insulin secretion is not stimulated in the absence of extracellular Ca2+ (Ca 0). |
OTHER PUTATIVE MESSENGERS IN THE β-CELL RESPONSE TO GLUCOSE
As previously discussed, changes in the cytoplasmic ATP/ADP ratio in β-cells are currently thought to be the major coupling factor between glucose metabolism and the biophysical events leading to the generation of the triggering signal. There is also evidence for a role of the ATP/ADP ratio in the amplification pathway (Fig. 6.2). Over the years, however, many other messengers have been considered to play a role in the β-cell secretory response to glucose.
Metabolic Factors
The hypothesis that a specific intermediate of glucose metabolism (e.g., of glycolysis) is directly involved in triggering secretion was abandoned when it became evident that fuel secretagogues metabolized through distinct pathways have largely similar effects.
Reduced pyridine nucleotides (NADPH and NADH) are unlikely to be an essential triggering signal but may play a modulatory role (e.g., by changing the thiol-disulfide equilibrium in various proteins) (107). Fuel-induced insulin secretion is associated with a more reduced state of the cytosol of β-cells (2,108,109).
The GTP/GDP ratio in islet cells increases in parallel with the ATP/ADP ratio upon stimulation with glucose (38,110). This parallelism probably reflects the high activity of the islet nucleotide-diphosphate kinase (111). There exists a good correlation between glucose-induced insulin secretion through either pathway and the GTP/GDP ratio (38), but a specific role of changes in GTP or GDP in stimulus-secretion coupling remains disputed (110,112).
A mitochondrial factor distinct from ATP has been suggested to control exocytosis of insulin granules together with Ca2+ and ATP. The proposal that this factor might be glutamate (113) has been challenged (114).
Malonyl Coenzyme A and Long-Chain Acyl Coenzyme A
The anaplerotic characteristics of glucose metabolism in β-cells lead to the formation of excess citrate that exits mitochondria to serve as a precursor of malonyl CoA in the cytoplasm (36) (Fig. 6.1). Malonyl CoA then inhibits carnitine palmitoyl transferase I and reduces fatty acid uptake by mitochondria, which explains the inhibition by glucose of the oxidation of endogenous (and exogenous) fatty acids (118,119). The result is an increase in cytosolic long-chain acyl CoAs that are proposed to serve as effector signal molecules (36). Because long-chain acyl CoAs open K+-ATP channels (120), they cannot underlie generation of a triggering signal by membrane depolarization. Long-chain acyl CoA seems to increase the efficacy of Ca2+ on exocytosis (121). This attractive model has, however, been challenged by the demonstration that glucose-induced insulin secretion does not correlate with the extent to which lipids are either oxidized or esterified (122). Other signaling functions of glucose-regulated anaplerosis are currently being investigated (123).
Cyclic Adenosine Monophosphate
Glucose increases the concentration of cAMP in β-cells slightly (124), probably secondarily to the increase of [Ca2+]i and the activation of adenylate cyclase by a Ca2+-calmodulin protein kinase (125). Several phosphodiesterases (PDE) that degrade cAMP are present in β-cells, but the PDE 3 isoform is predominant both quantitatively and functionally (126,127). There is no doubt that an increase in cAMP concentration in β-cells is not a sufficient signal to trigger insulin secretion, but it can amplify the response to various secretagogues. Whereas the role of this amplification during hormonal stimulation of β-cells is well established (see below), its role during stimulation by glucose alone is much less clear. Experiments with isolated β-cells suggest that a minimal concentration of cAMP is necessary for normal stimulation of insulin secretion by glucose (128), and hormones that increase cAMP levels have been suggested to increase the glucose competence of poorly responsive β-cells (129). In contrast, inhibitors of protein kinase A do not impair the effect of glucose in intact islets (130).
Inositol Phosphates and Diacylglycerol
Glucose stimulates both the synthesis and hydrolysis of phospholipids in β-cells (131,133,134). However, the breakdown of phosphatidylinositol 4,5-bisphosphate and accumulation of inositol 1,4,5-trisphosphate are not primary signals but rather consequences of the increase in [Ca2+]i (133,134). The acceleration of phospholipid turnover by glucose may serve to optimize the potentiation of insulin secretion by agonists acting via phospholipase C-linked receptors (see below).
Glucose stimulates de novo synthesis of diacylglycerol in β-cells. However, this diacylglycerol is richer in palmitate and poorer in arachidonate than is the diacylglycerol generated by hydrolysis of preexisting glycerolipids and does not significantly increase the total mass of diacylglycerol in islet cells (135). This may explain why glucose is usually (136,137,138), although not consistently (139,140), found not to translocate and activate protein kinase C in β-cells. It is possible that activation of protein kinase C (by diacylglycerol or another stimulator) contributes to the second phase of insulin secretion in certain species (102), but the bulk of the evidence indicates that its role in glucose-induced insulin secretion is not a major one.
Arachidonic Acid and Other Lipid Derivatives
Glucose stimulation of islet cells increases the release of arachidonic acid from phospholipids. However, decisive arguments that arachidonic acid itself plays a role in stimulus-secretion coupling are still missing (132,138,141). On the other hand, metabolites of arachidonic acid could modulate glucose-induced insulin secretion. The latter is decreased by prostaglandins and increased by certain products formed by lipoxygenation. It has been suggested that phosphatidic acid, formed by activation of phospholipases or by de novo synthesis from glucose, and lysophospholipids formed by activation of phospholipase A2, might be involved in glucose-induced insulin secretion (138). In no case, however, have the reported effects of these substances carried conviction that their role is critical in stimulus-secretion coupling.
GENERAL CHARACTERISTICS OF GLUCOSE-INDUCED INSULIN SECRETION
Links between Insulin Synthesis and Secretion
The mechanisms of insulin biosynthesis and of its regulation by glucose are dealt with in detail in Chapter 5.
Short-term control of insulin secretion does not depend on insulin synthesis. Pancreatic insulin stores largely exceed the maximal secretory rates (∼5% of insulin content per hour). The inhibition of insulin secretion that follows blockade of protein synthesis in β-cells does not reflect requirement of insulin synthesis (142) but results from the loss of other proteins with a short half-life (143). The first alteration is a rapid (1 to 2 hours) impairment of the action of Ca2+ on exocytosis followed by a delayed (>5 hours) decrease in the production of the triggering signal (143). Insulin synthesis and secretion can also be dissociated under several conditions. In contrast to glucose and other fuel secretagogues that stimulate both processes, several agents (e.g., arginine, acetylcholine, and hypoglycemic sulfonylureas) increase insulin secretion without increasing its synthesis (2,144). It also is possible to abolish the effects of glucose on secretion without affecting those on synthesis (e.g., by omitting extracellular Ca2+) (2,144).
Insulin synthesis and secretion are, however, not completely independent events. Under certain circumstances, isolated islets release newly synthesized insulin in preference to older stored hormone (145). This may indicate that insulin granules do not undergo exocytosis at random but that a subpopulation of newly formed granules is “marked” to be released rather than to be stored (146). The mechanisms of this marking are not known. Alternatively, the phenomenon of preferential release of newly formed insulin could be due to β-cell heterogeneity within the islets (147). If the rise in glucose concentration recruits the same β-cells to synthesize (148) and to release insulin, and if these cells have low stores of preformed insulin, an apparent preferential secretion of newly formed insulin may be measured.
Kinetics
Insulin secretion depends not only on the ambient concentration of glucose but also on the rate of change of this concentration. When the glucose level increases slowly, the rate of insulin secretion increases in parallel. However, when the concentration of glucose is abruptly increased and then maintained at a high level, insulin secretion follows a biphasic time course (Fig. 6.6C). A nadir and a slowly rising second phase follow a rapid peak (first phase). This pattern, first described in the perfused rat pancreas (149), is commonly observed in
vitro. It is not seen in vivo when the plasma glucose concentration progressively increases after a meal or even after an oral glucose load, but it can be produced by a rapid increase in plasma glucose during intravenous glucose infusion (150). This ability of β-cells to respond rapidly to glucose is thought to be essential for optimal glucose homeostasis. The loss of this ability has long been considered an early sign of β-cell dysfunction characteristic or even predictive of type 2 diabetes mellitus (150,151).
vitro. It is not seen in vivo when the plasma glucose concentration progressively increases after a meal or even after an oral glucose load, but it can be produced by a rapid increase in plasma glucose during intravenous glucose infusion (150). This ability of β-cells to respond rapidly to glucose is thought to be essential for optimal glucose homeostasis. The loss of this ability has long been considered an early sign of β-cell dysfunction characteristic or even predictive of type 2 diabetes mellitus (150,151).
Numerous hypotheses have been put forward to explain the biphasic time course of glucose-induced insulin secretion. Because the glucose dependency of the two phases is similar in vitro (152) and in vivo (150), there is no evidence that distinct initial steps in glucose recognition are involved. Early suggestions that the nadir between the two phases is due to a negative feedback exerted by insulin secreted during the first phase or to partial inhibition by somatostatin have been abandoned (152,153). The concept that the first phase reflects release of preformed insulin and that the second phase reflects release of newly synthesized insulin is also not correct (152).
According to a “storage-limited” model (152,153), two compartments of insulin granules could exist, the smaller one, in proximity to the plasma membrane, being readily released and responsible for the first phase. The existence of distinct subsets of granules is supported by experiments monitoring insulin release as capacitance changes in single, voltage-clamped β-cells (154,155). It has been suggested that the pool of readily releasable granules is composed of those insulin granules that can be immunoprecipitated by an antibody directed to syntaxin, a plasma membrane protein (156). However, functional evidence indicates that only a fraction of the docked granules is readily releasable (155). This model implies that the second phase reflects the energy-dependent mobilization of granules from a reserve to the readily releasable pool.
According to a “signal-limited” model (153,157), changes in the magnitude or effectiveness of the triggering signal produced by glucose could underlie the biphasic response. Evidence supporting this hypothesis is accumulating. The increase in β-cell [Ca2+]i produced by glucose is biphasic (74,75), and no first phase of secretion occurs when [Ca2+]i does not abruptly increase. However, no gradual increase in [Ca2+]i occurs during sustained glucose stimulation (74,75). Therefore, the slowly increasing second phase of secretion (which is more pronounced in the rat than in the mouse) (Fig. 6.6C) does not depend simply on the concentration of cytoplasmic Ca2+ but probably also involves an increase in the efficacy of the Ca2+ signal (42,102,158). There is good evidence that the amplifying pathway is implicated, but it is still unclear whether the enlargement of the pool of releasable insulin granules results from a mobilization of granules toward the sites of exocytosis or from a modification of the properties of already adequately situated granules.
Biphasic insulin secretion is not specific to glucose. It can also be induced by other fuel stimuli (159,160). Moreover, the kinetics of the secretory response may be influenced by the concentration of the applied stimulus and by the prevailing concentration of glucose. For instance, the common idea that sulfonylureas simply trigger a first phase of insulin secretion (149) holds true only for stimulation by high concentrations of the drugs in low-glucose environments. When used at a low concentration and in the presence of glucose, sulfonylureas induce a sustained secretion of insulin (161,162). These are further arguments supporting the conclusion that the kinetics of insulin secretion does not depend simply on the existence of different pools of insulin granules.
Either model is probably insufficient. Biphasic secretion of insulin requires immediately releasable and mobilizable granules and adequate temporal and quantitative changes in both triggering and amplifying signals. It is also unlikely that an empty pool of releasable granules can explain the lack of first phase in patients with type 2 diabetes mellitus (150,151). If this were the case, why should non-glucose stimuli cause immediate insulin secretion (163,164)? The defect is more likely due to the inability of glucose to induce a rapid increase in [Ca2+]i in these diseased β-cells.
Concentration Dependency: Recruitment and Increase in the Individual Responses
As previously mentioned, the relationship between the extracellular glucose concentration and the rate of insulin secretion in vitro is sigmoidal (1) (Fig. 6.6A). In isolated rat islets or in the perfused rat pancreas, the threshold concentration is around 5 to 6 mM and half-maximal and maximal responses are observed at 9 to 11 mM and 15 to 20 mM, respectively. This relationship is shifted slightly to the left in human islets and to the right in mouse islets, which corresponds well with the differences in blood glucose concentration between the three species. It is also important to bear in mind that circulating nutrients other than glucose, hormones, and neurotransmitters shift the dose-response relationship to glucose to the left in vivo (see below).
The sigmoidal shape of the dose-response curve has been attributed to a gaussian distribution of the thresholds for β-cell stimulation (128). Increasing the concentration of glucose would thus recruit more and more β-cells to secrete insulin. Functional heterogeneity between β-cells has been directly demonstrated in vitro by measuring (with the reverse hemolytic plaque assay) insulin secretion from single β-cells obtained by dispersion of rat islets (165). The number of secreting cells increases as the concentration of glucose is increased (166,167). The alternative possibility is that the response of each individual cell increases with the concentration of glucose. It has indeed been demonstrated that glucose causes a dose-dependent increase in [Ca2+]i (106) and in insulin release (166,167) in single isolated β-cells.
The key question is whether the heterogeneity of individual isolated β-cells persists within the islets of Langerhans in which β-cells are preferentially interconnected by gap junctions made of connexin 36 (168). This intercellular coupling and paracrine influences may erase the individual differences to constitute a functionally homogeneous population. Thus, in contrast to the heterogeneous responses produced in isolated β-cells, glucose induces uniform metabolic (NADPH autofluorescence) (169), electrical (69,71,170), and [Ca2+]i responses (74,75) in β-cells residing within intact islets. The homogeneity and synchrony of complex [Ca2+]i changes in different regions of an islet are illustrated in Figure 6.4. Recruitment of β-cells can be detected in islets or clusters, but it occurs over a narrow range of glucose concentrations (106). There is no doubt that the triggering signal is not generated in an all-or-none manner; its amplitude increases with the glucose concentration. It is also well established that the secretory performance is greatly improved when contacts are established between β-cells (128,171,172). Through the electrical coupling, the most active β-cells of an islet can entrain the less active ones to generate an increase in [Ca2+]i. Nevertheless, there is indirect evidence that not all β-cells within intact islets secrete insulin at the same rate (173,174). This heterogeneity of secretion in face of a homogeneous triggering signal could be due to a variable production of the amplifying signals (106).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


