Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .




Introduction
Catecholamines are monoamine neurotransmitters and circulating hormones derived from the amino acid tyrosine, produced by the sympathetic nervous system, brain, and adrenal medulla [ ]. The three physiologically active catecholamines synthesized in the human body are epinephrine (adrenaline), norepinephrine (noradrenaline), and
Catecholamines and autonomic physiology
The synthesis, storage, and regulation of catecholamines
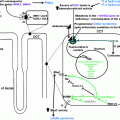
Catecholamines contain a catechol (1,2-dihydroxy benzene) moiety, an ethylamine side chain, and an amine group with additional substitutions [ ]. The chemical structure of the catecholamines, the steps involved in the catecholamine biosynthetic pathway, and the enzymes involved are depicted in Fig. 2.1 . The first step incatecholamine biosynthesis is the conversion of phenylalanine to tyrosine. Tyrosine is further converted to l -DOPA ( l -3,4-dihydroxyphenylalanine) by tyrosine hydroxylase, the rate-limiting step of catecholamine biosynthesis. This is followed by decarboxylation of l -DOPA into dopamine by the enzyme l -aromatic amino acid decarboxylase. The pathway stops here in the dopaminergic neurons located in the midbrain nuclei, namely the ventral tegmental area and the substantia nigra pars compacta [ ].
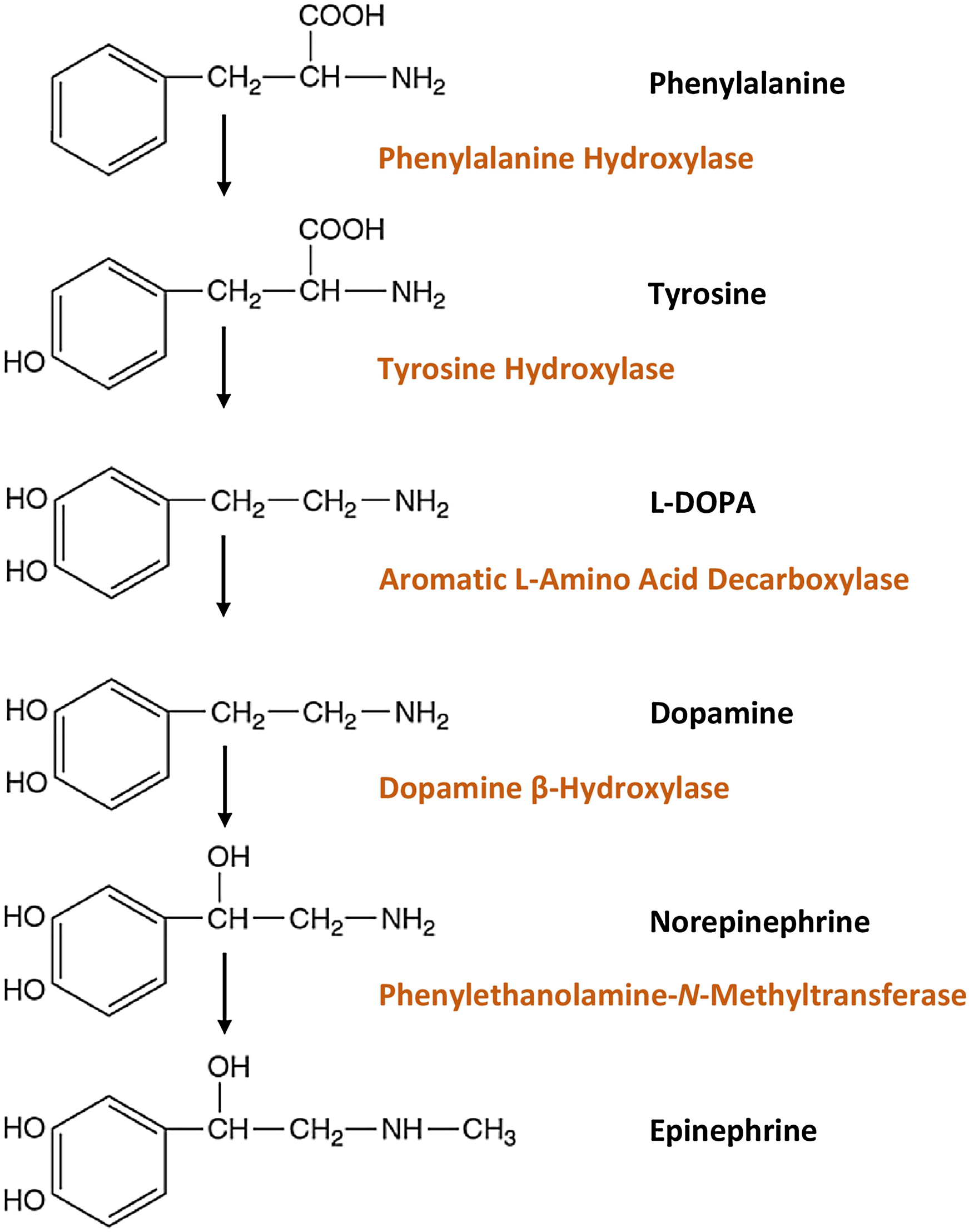
In the postganglionic neurons of the sympathetic nervous system and in the clusters of noradrenergic neurons (the largest cluster being the locus coeruleus) located in the pons and the medulla, further hydroxylation of dopamine by the enzyme dopamine β-hydroxylase (DBH) leads to formation of norepinephrine [ ]. Subsequent N-methylation of norepinephrine yields epinephrine [ ]. This process is catalyzed by the enzyme phenylethanolamine-N-methyltransferase (PNMT) which uses S-adenosyl- l -methionine as the methyl donor. This conversion of norepinephrine to epinephrine occurs within chromaffin cells of the adrenal medulla, neurons of the rostral ventrolateral medulla (RVLM), and neurons of the nucleus tractus solitarius (NTS).
Sympathetic neurons, unlike chromaffin cells within the adrenal medulla, do not possess PNMT and therefore cannot synthesize epinephrine [ ]. Therefore, the adrenal medulla is the major source of epinephrine released into systemic circulation. The adrenal medulla releases predominantly epinephriine (80%), followed by norepinephrine (19%) and dopamine (1%). The function of PNMT is potentiated by the presence of glucocorticoids, which are produced and released by the adrenal cortex, creating a physiologic link between the adrenal cortex, medulla, and the release of epinephrine [ ]. The organ of Zuckerkandl (chromaffin cells located around the aorta from the level of the superior mesenteric artery to the level of the aortic bifurcation), neurons of the amygdala, and neurons of the retina also express PNMT and can produce epinephrine [ ].
The RVLM is one of the most important regions involved in the maintenance of sympathetic tone [ ]. The RVLM sends direct monosynaptic projections to the intermediolateral columns of the thoracolumbar spinal cord (from T1 to L2) where the sympathetic preganglionic neurons are located.
In the postganglionic neurons of the sympathetic nervous system, norepinephrine is stored in vesicles and released into the synaptic cleft by exocytosis. This exocytosis is prompted by acetylcholine released by preganglionic neurons—which are themselves stimulated to do so [ , ]. The norepinephrine that is released from these vesicles binds to postsynaptic adrenoceptors present on either target cells or on presynaptic adrenoceptors to modulate the presynaptic neuronal activity [ ]. Norepinephrine also undergoes reuptake either into the noradrenergic neurons or into the target cells through the norepinephrine transporter (NET). Nearly 80%–95% of norepinephrine that is released from vesicles will undergo active reuptake into noradrenergic neurons [ ]. This reuptake subsequently returns norepinephrine to storage vesicles. Ultimately, only a minority of norepinephrine binds to corresponding adrenoceptors [ ].
In chromaffin cells of the adrenal medulla, catecholamines are stored in vesicles and released into the bloodstream by exocytosis [ ]. Exocytosis of these catecholamines occurs when acetylcholine released by preganglionic sympathetic nerve fibers binds to nicotinic cholinergic receptors present on chromaffin cells, leading to pursuant depolarization of chromaffin cells which activates voltage-gated calcium channels. The consequent rise in intracellular calcium leads to vesicular exocytosis. Upon release, these circulating catecholamines bind to their cognate adrenoceptors on target organs (discussed below). Notably, epinephrine and norepinephrine have short half-lives due to their rapid metabolism into inactive products thus leading to often short-lived physiologic responses.
Metabolism of catecholamines
Under physiologic conditions catecholamines are metabolized mainly by catechol-O-methyltransferase (COMT, COMT-MB and COMT-S for membrane bound and soluble COMT enzymes, respectively) and monoamine oxidase (MAO-A and MAO-B) [ ]. Catecholamine metabolism is depicted in Fig. 2.2 . COMT catalyzes the O-methylation of dopamine into methoxytyramine, norepinephrine into normetanephrine, and epinephrine into metanephrine. MAO catalyzes the oxidative deamination of dopamine into 3,4 dihydroxyphenylacetic acid (DOPAC), and norepinephrine and epinephrine into 3,4 dihydroxyphenylglycol (DHPG). The metabolites catalyzed by MAO can be metabolized further by COMT, and those metabolites catalyzed by COMT can be metabolized further by MAO. The final product of dopamine metabolism is homovanillic acid, and that of norepinephrine and epinephrine metabolism is vanillic acid.
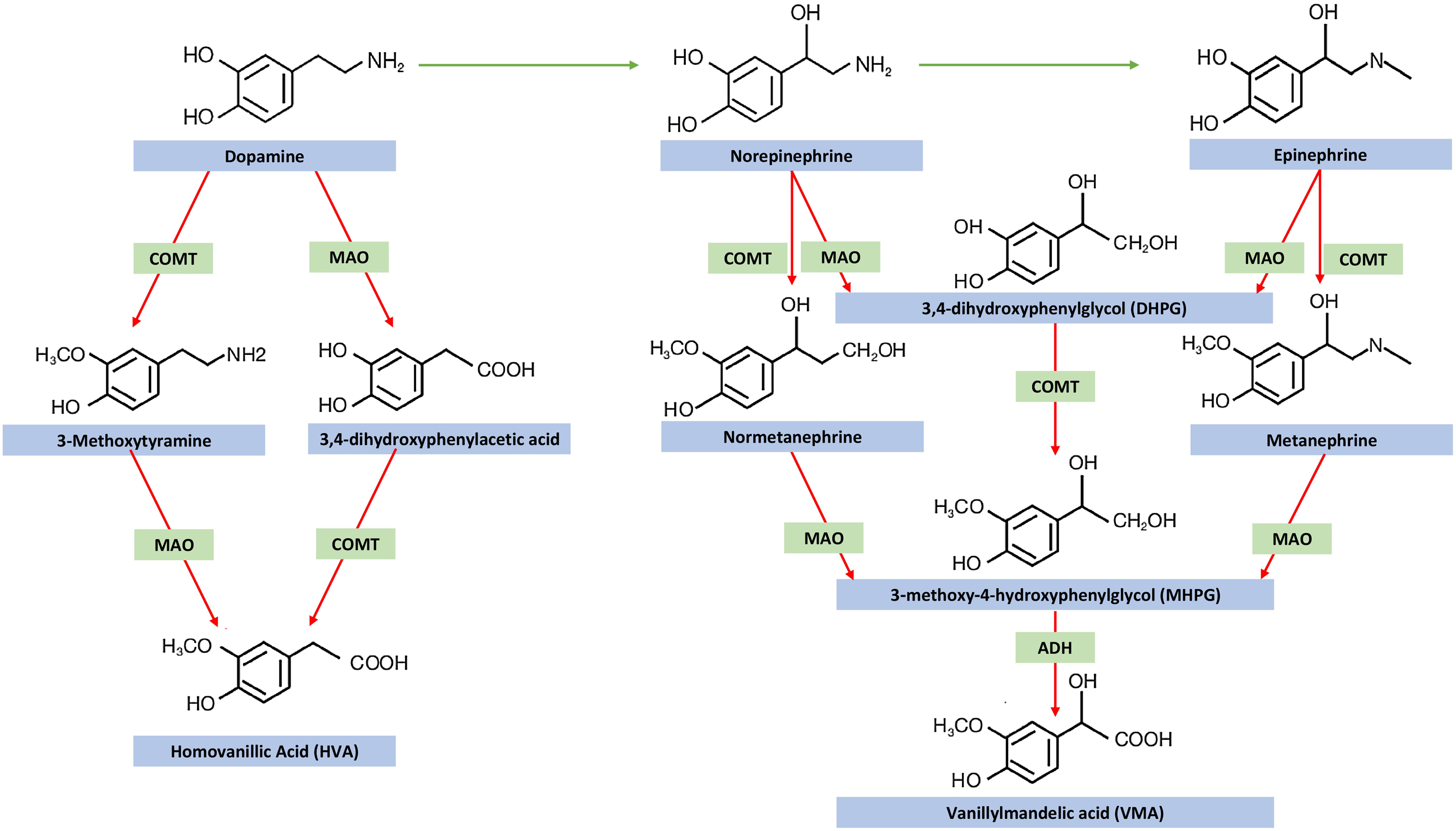
COMT is absent in sympathetic nerves; therefore, norepinephrine in sympathetic nerves is metabolized by deamination rather than O-methylation [ ]. On the other hand, due to the presence of COMT within the cytoplasm of the adrenal medullary and extraadrenal chromaffin cells, O-methylation metabolites are relatively specific to chromaffin cells. In normal individuals, at least 90% of metanephrine and nearly 25% of normetanephrine are produced within adrenal chromaffin cells. Of the remaining 75% of normetanephrine, nearly 15% is derived from extraadrenal chromaffin cells, while 60% is derived from the metabolism (by extraneuronal cells) of norepinephrine released by sympathetic nerves.
Adrenoceptors, subtypes, and associated second messenger systems
Adrenoceptors are G-protein-coupled receptors (GPCRs) which are composed of (1) an extracellular element that binds catecholamines, (2) seven transmembrane domains (which anchor the protein to the cell membrane), and (3) an intracellular element that allows the bound receptor to interact with the G-protein present within the cell [ ]. This structure lends to the functional ability to transduce an extracellular signal into an intracellular signal. Functionally, a catecholamine (the ligand) binds to the GPCR resulting in a conformational change, causing an interaction with a G-protein. This G-protein in turn then modulates the activity of the enzyme attached to the G-protein and it is these enzymes and their downstream effectors that define canonical second messenger pathways (elaborated upon below). These G-proteins are composed of three distinct subunits: α, β, and γ. It is the α subunit that confers the name of the G-protein. Accordingly, the G s protein has a stimulatory α subunit that activates the enzyme adenylyl-cyclase; the G i protein has an inhibitory α subunit that inhibits the enzyme adenylyl-cyclase; and the G q protein has a stimulatory α subunit that activates the enzyme phospholipase C [ ]. The physiology of the GPCR signaling pathway is depicted in Fig. 2.3 .
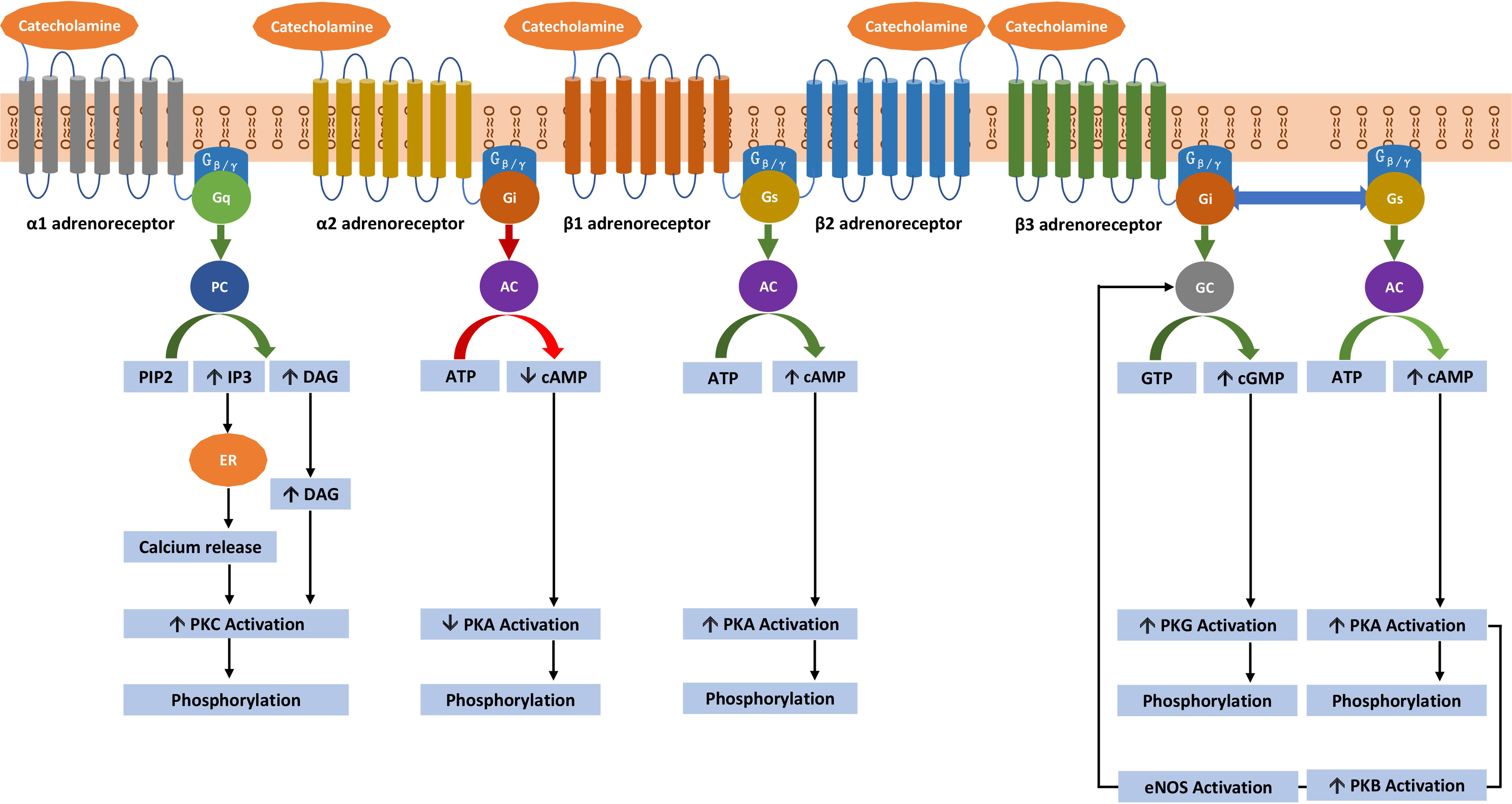
Adrenoceptors are divided into α 1 , α 2 , β 1 , β 2 , and β 3 receptor subtypes. The α 1 -adrenoceptors are subdivided into three subtypes namely α 1A , α 1B , and α 1D [ ]. Similarly, the α 2 -adrenoceptors have at least three subtypes namely α 2A , α 2B , and α 2C [ ]. However, this text will focus on the major functions of α 1 – and α 2 -adrenoceptors. α 1 -adrenoceptor stimulation of vascular smooth muscle cells, present within blood vessels, results in their contraction. This contraction of smooth muscle cells, oriented radially within the tunica media of arteries and veins, leads to inward narrowing of the vascular lumen or vasoconstriction. Moreover, α 1 -adrenoceptor stimulation of the heart mediates positive inotropic effects and coronary artery vasoconstriction. On the other hand, α 2 -adrenoceptor stimulation within the central nervous system (presynaptic neurons) reduces sympathetic outflow and consequent sympathetic tone (by inhibiting the release of norepinephrine). This results in a decrease in blood pressure [ ]. α 2 -adrenoceptor stimulation also causes coronary vasoconstriction.
The stimulation of β 1 -adrenoceptors in the heart (which account for nearly 80% of the β-adrenoceptors of the heart) results in a positive inotropic (increased force of contraction), chronotropic (increased heart rate), dromotropic (increased electrical conduction velocity), and lusitropic (increased ventricular relaxation) effects [ ]. In addition, stimulation of β 1 -adrenoceptors mediates coronary artery vasodilatation and increases automaticity while decreasing refractory period within cardiomyocytes. In the juxtaglomerular cells of the kidneys, β 1 -adrenoceptor stimulation mediates renin release.
The stimulation of β 2 -adrenoceptors within vascular smooth muscle cells results in radial relaxation of blood vessels and resultant peripheral arterial and venous vasodilatation [ ]. β 2 -adrenoceptor stimulation also affects bronchodilatation and relaxation of muscles of the uterus (tocolytic effect), urinary bladder, and gastrointestinal tract. The stimulation of β 2 -adrenoceptors in the heart (which accounts for nearly 20% of the β-adrenoceptors of the heart) results in positive inotropic and chronotropic effects with enhanced lusitropy. In addition, it increases the automaticity and decreases the refractory period within cardiomyocytes and mediates coronary artery vasodilatation.
The effect of adrenoceptor stimulation on a particular vascular bed or blood vessel is determined by the distribution of adrenoceptors and the affinities of those adrenoceptors for epinephrine and norepinephrine. β 2 -adrenoceptors are present on blood vessels feeding skeletal muscles, whereas α 1 -adrenoceptors are present on metarterioles [ , ]. At high concentrations, epinephrine acts on both β 2 – and α 1 -adrenoceptors. Through this mechanism, it can cause a decrease in diastolic blood pressure (mediated through β 2 -adrenoceptor stimulation), but an increase in systolic blood pressure (mediated through α 1 -adrenoceptor stimulation).
Though both epinephrine and norepinephrine can bind to and stimulate β 1 -adrenoceptors, the tendency to cause a tachyarrhythmia in catecholamine excess states differs between these two catecholamines [ ]. Epinephrine has a greater tendency to cause tachyarrhythmias given its relatively high affinity for β 1 – compared to α 1 -adrenoceptors. On the contrary, norepinephrine has less of a tendency to cause tachyarrhythmias and a greater tendency to cause hypertension due to its high affinity for α 1 – compared to β 1 -adrenoceptors. Further, in some instances, epinephrine may lead to hypotension due to excessive β 2 -adrenoceptor-mediated vasodilation. This phenomenon can be seen in epinephrine secreting pheochromocytomas and paragangliomas (PPGLs) and during preoperative treatment of pheochromocytomas with sole α 1 -adrenoceptor blockade due to a relative excess of catecholamines available to bind β 2 -adrenoceptors.
The β 3 -adrenoceptors are most abundant in white and brown adipose tissue, where they mediate lipolysis, fatty acid oxidation, and thermogenesis [ ]. In the cardiovascular system, the β 3 -adrenoceptors are present in the cardiomyocytes where they modulate cardiac function (positive lusitropy—with improved diastolic function and negative inotropy—with decreased myocardial oxygen consumption), and in the vasculature where they modulate vasodilatation and angiogenesis—with enhanced oxygen supply. β 3 -adrenoceptor stimulation requires a higher catecholamine concentration (at the receptor level) than is required for β 1 – and β 2 -adrenoceptors activation, indicating that it could act as a brake to prevent β 1 – and β 2 -adrenoceptor overactivation. β 3 -adrenoceptors are an “up and coming” area of research in cardiac function and heart failure but they are still being researched. The β 3 -adrenoceptor response to catecholamine stimulation seems to be preserved even after prolonged catecholamine stimulation. On the other hand, the β 1 – and β 2 -adrenoceptor responses are diminished by excessive stimulation leading to desensitization.
In cardiomyocytes, the desensitization to sustained β 1 – and β 2 -adrenoceptor stimulation is mediated by adrenoceptor phosphorylation via G-protein-coupled receptor kinases (GRKs), followed by binding to β-arrestin2 [ , ]. The GRK2/β-arrestin2 complex facilitates uncoupling of G-proteins from their respective β 1 – and β 2 -adrenoceptors leading to internalization of β 1 -and β 2 -adrenoceptors. In addition, a switch from G s to G i protein occurs in β 2 – and not β 1 -adrenoreceptors [ ]. The coupling of β 2 -adrenoceptors to the G i protein results in decreased cardiac contractility. Moreover, the switch happens only with epinephrine excess, not with norepinephrine excess (given differing affinities for the β 2 -adrenoceptor). Epinephrine activates both the G s and G i pathways (G s at lower and G i at higher concentrations), whereas norepinephrine activates only the G s pathway through the β 2 -adrenoceptor. Furthermore, norepinephrine exhibits a 20-fold lower affinity toward β 2 -adrenoceptors compared to β 1 -adrenoceptors. This difference in affinity likely explains why epinephrine-secreting PPGLs are commonly associated with catecholamine-induced stress cardiomyopathy in comparison to norepinephrine-secreting and dopamine-secreting PPGLs.
There are at least five dopamine receptors (D1–D5), which are GPCRs that are divided into two subfamilies: the D1-like receptor subfamily that corresponds to the original D1 dopamine receptor is comprised of D1 and D5 receptors and the D2-like receptor subfamily corresponds to the original D2 receptor and is comprised of D2, D3, and D4 receptors [ ]. The D1-like subfamily of dopamine receptors is coupled to the G s protein that activates adenylyl-cyclase, which increases the generation of the second messenger cyclic adenosine monophosphate (cAMP), activating protein kinase A (PKA) leading to phosphorylation of downstream proteins. On the other hand, the D2-like subfamily of dopamine receptors is coupled to the G i protein that inhibits adenylyl-cyclase, decreasing the generation of cAMP and PKA-mediated phosphorylation of downstream proteins.
Neuroendocrine effects of catecholamines on blood pressure
Ultimately, a simple signal, a rise in a given catecholamine, can lead to a complex coordinated response from the cardiovascular system. Adrenoceptors modulate cell function by influencing intracellular second messenger systems. This occurs because of adrenoceptor subtypes, each with differing affinities for catecholamines. These adrenoceptor subtypes are themselves present with varying levels of expression and differing spatial densities on end-organs (namely throughout the vasculature and the heart). This system ramifies a binary signal into multiple different subsignals all acting together with one unifying prerogative, to stimulate cardiovascular performance. In this way, a rise in epinephrine may enact vasoconstriction of certain vessels via α 1 -adrenoceptor stimulation, while paradoxically leading to vasodilation of other vessels due to stimulation of β 2 -adrenoceptors, and finally augmenting cardiac output via its effect on β 1 – and β 2 -adrenoceptors. Though seemingly haphazard, these functions unify to direct an increase in cardiac output (mediated by β 1 – and β 2 -adrenoceptor stimulation) toward vital tissues (for example, via β 2 -adrenoceptor-mediated vasodilation) and away from nonessential tissues (for example, via α 1 -adrenoceptor-mediated vasoconstriction). Thus, to fully understand the effects of catecholamines upon blood pressure and cardiac performance, one must be familiar with the predominant adrenoceptor present within a given vascular bed or blood vessel, its paired second messenger system, and its final effect on downstream effectors, namely ion channels and the actin–myosin cross-bridge. These relationships are elaborated upon in Fig. 2.3 and are discussed below [ ].
Different vascular beds or blood vessels have varying expression of adrenoceptors—allowing each to act in a specific manner in response to a given catecholamine. However, three major patterns exist regarding adrenoceptor expression throughout the vasculature and consequent effects on blood pressure. Stimulation of α 1 -adrenoceptors on metarterioles leads to vasoconstriction and is a major determinant of increasing blood pressure. β 2 -adrenoceptor stimulation on blood vessels feeding skeletal muscle can allow for improved blood flow in times of stress; however, excessive stimulation may lead to a decrease in blood pressure. Finally, α 2 -adrenoceptor expression on presynaptic terminals establishes a negative feedback pathway. Stimulation of α 2 -adrenoceptors may then blunt the release of norepinephrine (for example at metarterioles) which can lead to a decrease in blood pressure.
The α 1 -adrenoceptors present upon vascular smooth muscle cells are coupled to the G q protein that activates the enzyme phospholipase C, which cleaves phosphatidylinositol 4,5-bisphosphate yielding inositol triphosphate (IP3) and diacylglycerol [ ]. Among other pathways, IP3 binds to its cognate receptor on the endoplasmic reticulum leading to a release of calcium into the cytoplasm. Calcium then augments actin–myosin cross-bridge cycling by leading to downstream phosphorylation of myosin. Increased actin–myosin cross-bridge cycling leads to sarcomere contraction at metarterioles on precapillary sphincters, increasing total peripheral resistance. By maintaining more blood within the arterial compartment, this leads to a rise in blood pressure. The net result of α 1 -adrenoceptor activation by norepinephrine or epinephrine is vasoconstriction and hypertension.
Stimulation of α 2 -adrenoceptors on presynaptic terminals, via catecholamines, defines a negative feedback pathway [ ]. The α 2 -adrenoceptor stimulation inhibits adenylyl-cyclase via the G i protein; this leads to a decrease in cAMP, in turn, reducing the stimulation of PKA leading to less vesicular exocytosis of catecholamines. In this way, α 1 -adrenoceptor stimulation via canonical second messengers increases the blood pressure, while α 2 -adrenoceptor stimulation via a negative feedback pathway decreases the blood pressure.
The β 2 -adrenoceptors present on vascular smooth muscle and the heart are coupled to the G s protein. When stimulated by epinephrine, the coupled G s protein increases cAMP levels in both cardiomyocytes and vascular smooth muscle cells. In vascular smooth muscle cells, the rise in cAMP leads to downstream inhibition of myosin light chain kinase (MLCK) [ ]. This action on MLCK reduces phosphorylation of myosin and reduces actin–myosin cross-bridge cycling leading to vasodilation and increasing blood flow to skeletal muscle. When epinephrine levels are elevated, as previously mentioned, excessive vasodilation can lead to a drop in diastolic blood pressure. On the contrary, in cardiomyocytes, the G s -mediated rise in cAMP activates MLCK with resultant phosphorylation of myosin; this increases the force of myocardial contraction [ ].
Within the heart, β 1 – and β 2 -adrenoceptors are present on cardiac conductive cells within the sinoatrial node (SA Node) and the atrioventricular node (AV Node) as well as the atrial- and ventricular cardiomyocytes. β 1 -adrenoceptors are heavily expressed upon conductive cells of the SA and AV node and their stimulation has a dromotropic and chronotropic effect. Within the SA node, stimulation of β 1 -adrenoceptors by catecholamines leads to a rise in cAMP which stimulates the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel leading to an influx of positive ions augmenting the rate of spontaneous depolarization and therefore the heart rate (increased chronotropy) [ ]. Within the AV node, stimulation of β 1 -adrenoceptors by catecholamines leads to a rise in cAMP, stimulating PKA and leading to phosphorylation of L-type voltage-gated calcium channels (L-type VGCCs). This leads to a rise in intracytoplasmic calcium leading to more brisk depolarization and a quickening of conduction velocity through the atrioventricular node (increased dromotropy). Regarding force of contraction, catecholamines increase inotropy by binding to and stimulating β 2 -adrenoceptors present upon cardiomyocytes, while enhancing lusitropy via β 1 and β 2 -adrenoceptors. This leads to a rise in cAMP and PKA-mediated phosphorylation of L-type VGCCs, the pursuant influx of calcium augments inotropy [ , ]. Finally, both β 1 – and β 2 -adrenoceptor stimulation on ventricular cardiomyocytes increases ventricular relaxation (lusitropy) allowing for enhanced diastolic filling, which upon contraction leads to increased ejection fraction and forward delivery of blood into the arterial system [ ].
The β 3 -adrenoceptor is coupled to both the G s and G i protein [ ]. β 3 -adrenoceptor-mediated stimulation via G s activates both adenylyl-cyclase and guanylyl-cyclase. This enhances the generation of cAMP and cyclic guanosine monophosphate (cGMP) which, in turn, activates PKA and protein kinase G (PKG), respectively. On the other hand, the β 3 -adrenoceptor-mediated G i pathway stimulation activates guanylyl-cyclase, generating cGMP, and activating PKG in isolation. Both PKA and PKG stimulate the phosphorylation of the myofilament components like cTnI and calcium handling proteins like phospholamban. In addition, PKA phosphorylates and activates L-type VGCCs, whereas PKG inactivates them. The β 3 -adrenoceptor-mediated G s pathway stimulation with resultant PKA activation leads to the activation of protein kinase B (PKB) and endothelial nitric oxide synthase (eNOS) leading to nitric oxide generation. Nitric oxide generation can occur via enhanced eNOS mediated by both the G s and G i pathway, and from enhanced neuronal nitric oxide synthase via the G i pathway. The rise in nitric oxide levels, mediated by β 3 -adrenoceptor stimulation, enhances myocardial lusitropy and reduces myocardial inotropy. Thus, targeting β 3 -adrenoceptors could provide a novel approach to improving cardiac function, metabolism, and deleterious cardiac remodeling [ ].
Metabolic regulation by catecholamines
Catecholamines, predominantly epinephrine, less so norepinephrine, cause a hypermetabolic state with significant insulin resistance [ ]. Catecholamines stimulate aerobic glycolysis, glycogenolysis, and gluconeogenesis, and inhibit glycogen synthesis. They stimulate lipolysis, ketogenesis, and proteolysis to provide sufficient glucose precursor molecules, such as fatty acids and alanine. These together will result in hyperglycemia and hyperlactatemia. In the mitochondria, they promote mitochondrial uncoupling, and aggravate oxidative stress either through an accelerated glycolytic pathway, or through catecholamine autooxidation, thereby contributing to mitochondrial dysfunction. Catecholamines lower the potassium level due to β 2 -adrenoceptor activation which stimulates potassium uptake into cells.
Biologic effects
Summarizing the net effect of catecholamines on vascular hemodynamics
As previously mentioned, the physiologic effects of catecholamines depend on the concentration of a catecholamine, the tissue distribution of adrenoceptors, and differences in affinity toward adrenoceptors [ ]. These cardiovascular effects of catecholamines are summarized in Table 2.1 . In blood vessels, at low epinephrine levels, preferential activation of β-adrenoceptors—especially the β 2 -adrenoceptors—mediates a hypotensive response. On the other hand, at high epinephrine levels, the preferential activation of α-adrenoceptors (especially α 1 -adrenoceptors) mediates a hypertensive response. In the heart, the epinephrine-mediated β 1 -adrenoceptor stimulation results in a positive inotropic and chronotropic effect, whereas the epinephrine-mediated β 2 -adrenoceptor stimulation results in improved coronary circulation. This improved coronary circulation persists even at higher epinephrine levels despite greater α 1 -adrenoceptor mediated coronary artery vasoconstriction. This is because coronary blood flow is enhanced due to an increased relative duration of diastole and through local release of vasodilators from adrenoceptor-stimulated myocardium. These effects largely counterbalance the direct α 1 -adrenoceptor-mediated increase in coronary arterial tone.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


