(1)
Department of Surgery, Dar A lAlafia Medical Company, Qatif, Saudi Arabia
14.1 Introduction
Sickle cell anemia is an autosomal recessive disorder that results from a change of a single amino acid in the beta chain of hemoglobin.
In sickle cell anemia, the amino acid glutamic acid at the 6th position of the 146 amino acids of the beta chain of hemoglobin is replaced by valine.
Although this single change of one amino acid is the underlying defect of sickle cell anemia, it does not explain the heterogeneity of SCA that is observed clinically.
In the recent years, two sub-phenotypes of SCA have been described.
The “viscosity-vaso-occlusion” sub-phenotype. This is responsible for the erythrocyte sickling related complications such as:
Painful vaso-occlusive crisis
Acute chest syndrome
Avascular necrosis
The “hemolysis-endothelial dysfunction” sub-phenotype. This is responsible for the sickle cell anemia vasculopathy that involves:
Pulmonary hypertension
Priapism
Leg ulcers
Sudden death
Stroke
Asthma
In the clinical setting, there may be an overlap between these two sub-types.
This, however, is of clinical and therapeutic importance and may help when considering therapies for a specific sub-phenotype.
The pathophysiology of sickle cell anemia is complex, and several factors may play a role.
Recently, it was shown that dysfunction of the vascular endothelium in these patients plays a major role in the pathophysiology of sickle cell anemia, and impaired nitric oxide (NO) bioavailability represents the central feature of endothelial dysfunction.
NO is an endogenous vasodilator.
It is synthesized by endothelial cells from L-arginine, which is converted to citrulline by the NO synthases enzymes.
It is well known that NO generated by healthy endothelial cells has several functions:
Vasorelaxant
Anti-inflammatory
Antithrombotic
Deficiency of NO in patients with SCA is an important factor in the pathogenesis of SCA vasculopathy.
Decreased NO bioavailability has several adverse effects including:
Endothelial cell activation
Upregulation of the potent vasoconstrictor endothelin-1
Vasoconstriction
Platelet activation
Increased tissue factor
Activation of coagulation
All of these contribute to the clinical manifestations seen in patients with sickle cell anemia.
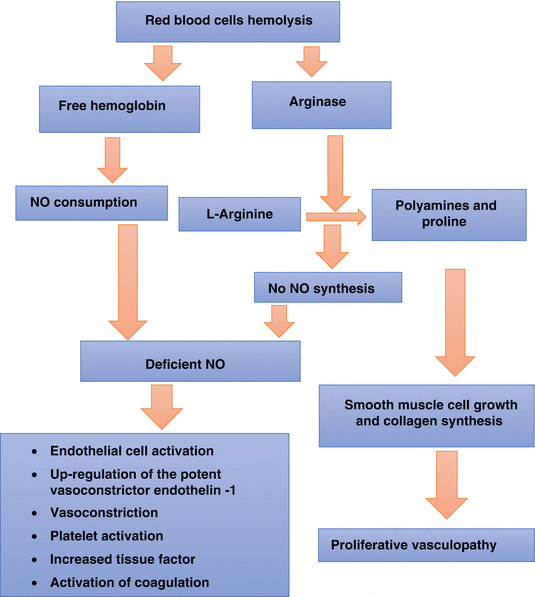
Pathophysiology of sickle cell vasculopathy:
Normally, the hemoglobin is stored in the red blood cells, but when hemolysis occurs, it is released into the plasma where it rapidly reacts with and destroys NO.
It is the heme part of the hemoglobin that reacts with NO leading to its consumption.
Add to this the NO scavenging by vascular superoxide anion.
NO consumption and the formation of reactive oxygen species ultimately inhibit vasodilatation.
Red blood cell hemolysis will also lead to the simultaneous release of erythrocyte arginase.
This will limit the availability of arginine to NO synthase to form NO, contributing also to the deficiency of NO.
Arginase will redirect the metabolism of arginine away from NO to ornithine and the formation of polyamines and proline, which are essential for smooth muscle cell growth and collagen synthesis.
With the shift away from NO toward ornithine metabolism, arginase contributes to the proliferative vasculopathy common to sickle cell anemia, pulmonary hypertension, and cardiovascular disease.
The levels of asymmetric dimethylarginine (an arginine analog) are also elevated in SCA, associated with hemolysis, pulmonary hypertension, and mortality, and will also affect the overall global arginine bioavailability.
Low arginine bioavailability may further compromise NO generation due to NO synthase uncoupling.
NO inhibits platelet activation, tissue factor expression, and thrombin generation, and intravascular hemolysis has the potential to drive a procoagulant state in these patients.
Hemolysis in patients with SCA leads to deficiency of NO for the following reasons:
NO scavenging by the heme of sickle hemoglobin released into the plasma by the hemolyzed RBCs
NO scavenging by vascular superoxide anion
Depletion of plasma arginine by arginase released by lysed RBCs
Endogenous NO synthesis inhibitors
Inactivation of tetrahydrobiopterin, a NO cofactor leading to the production of superoxide anion rather than NO
14.2 Pulmonary Artery Hypertension (PAH)
The normal pulmonary artery pressure is 15 mmHg.
PAH is defined as mean pulmonary artery pressure > 25 mmHg at rest or > 30 mmHg during exercise.
There are several causes for pulmonary artery hypertension which include primary and secondary causes.
The exact cause of primary PAH is not known.
Secondary PAH can be caused by several conditions including:
Chronic lung disease and heart disease
Collagen vascular disease
Thromboembolism
HIV
Liver cirrhosis
Anorexic agents
Sickle cell anemia
14.3 Pathophysiology of PAH
Intravascular hemolysis leading to NO deficiency is considered the main etiological factor of PAH in patients with SCA.
A relative deficiency of NO will lead to vasoconstriction and eventual remodeling of pulmonary vessels by smooth muscle cell growth and collagen synthesis.
Add to this platelet aggregation and attachment, release of growth factors, and procoagulant factors.
Thickening of the media and intima of small pulmonary vessels.
Medial thickening of muscular arteries and extension of muscle cells into nonmuscular arterioles.
Thickening, concentric non-laminar intimal hyperplasia, and fibrosis.
Pulmonary artery hypertension (PAH) is a relatively common and severe complication of sickle cell anemia.
PAH is also an independent risk factor for mortality in patients with sickle cell anemia.
Patients with SCA are known to have an anemia secondary to hemolysis. This will lead to an increase in the cardiac out.
This increase in cardiac output requires precapillary pulmonary microcirculation vasodilatation in order to accommodate the increased flow of blood.
Failure of this vasodilatation for any reason will result in an increase in pulmonary pressure.
It was shown that patients with SCA have high pulmonary pressures and pulmonary vascular resistance.
Patients with SCA and pulmonary hypertension have a significantly increased mortality rate when compared with SCA patients without pulmonary hypertension.
A mortality as high as 40 % was reported in SCA patients with pulmonary hypertension when compared to those without pulmonary hypertension.
The etiology of PAH in patients with SCA is multifactorial, and several mechanisms have been proposed including:
Intravascular hemolysis leading to nitric oxide (NO) deficiency
Progressive interstitial pulmonary fibrosis secondary to acute chest syndrome
Pulmonary vasculopathy secondary to platelet activation
Pulmonary vascular intimal thickening
Hypoperfusion of the pulmonary vascular bed
Recurrent thromboembolic disease and in situ pulmonary thrombosis
The prevalence of PAH in SCA patients increases with age, and it is associated with a high risk for death.
The high prevalence is also attributed to the fact that with improved care and new modalities of treatment, more children and young adults with SCA are surviving to an older age group and developing this complication later.
The risk factors for PAH in patients with SCA include:
Older age
Degree of hemolysis
Increased blood pressure
History of renal or heart disease
Iron overload
CholestasisRelated posts:
 Recent Advances in the Treatment of Sickle Cell Anemia
Hydroxyurea Treatment for Sickle Cell Anemia
Recent Advances in the Treatment of Sickle Cell Anemia
Hydroxyurea Treatment for Sickle Cell Anemia
 Perioperative Management of Patients with Sickle Cell Anemia
Perioperative Management of Patients with Sickle Cell Anemia
 Acute Appendicitis and Sickle Cell Anemia
Acute Appendicitis and Sickle Cell Anemia
 Genetics and Pathophysiology of Sickle Cell Anemia
Genetics and Pathophysiology of Sickle Cell Anemia
 Hepatobiliary Complications of Sickle Cell Anemia
Hepatobiliary Complications of Sickle Cell Anemia

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


