Until recently, an understanding of the molecular basis of NETs has been elusive. Banck et al.10 performed exome sequencing on 48 small intestine NETs. They reported an average of 0.1 somatic single nucleotide variants per 106 nucleotides (range, 0 to 0.59), mostly transitions (C>T and A>G). They discovered 197 protein-altering somatic single nucleotide variants affected mostly cancer genes, including FGFR2, MEN1, HOOK3, EZH2, MLF1, CARD11, VHL, NONO, and SMAD1. Alterations with potential therapeutic application were found in 35 patients, including SRC, SMAD family genes, AURKA, EGFR, HSP90, and PDGFR. Mutually exclusive amplification of AKT1 or AKT2 was the most common event in the 16 patients with alterations of phosphatidylinositol-3 kinase/protein kinase B/mammalian target of rapamycin (mTOR) signaling.10
Screening
The low NET incidence rates do not warrant population screening. For certain patients with an inherited predisposition for NETs (like in multiple endocrine neoplasia [MEN] syndromes), screening programs are often instituted. See Chapter 87 on MEN.
GENERAL PRINCIPLES OF NEUROENDOCRINE TUMOR DIAGNOSIS, STAGING, AND MANAGEMENT
General Diagnostic Principles
The diagnosis of GI and pulmonary NETs is often incidental, though patients with functional tumors can present with symptoms of hormone excess. The diagnostic approach for a patient with NETs is summarized by the North American Neuroendocrine Tumor Society11 and the National Comprehensive Cancer Network NET guidelines.12
Serum hormone markers are often elevated in NETs and can be a surrogate marker of symptoms of hormone excess or tumor growth, though none are sensitive enough to be used as a screening test. Chromogranin A levels are elevated in approximately 80% of patients with GI NETs; sensitivity is 75% and specificity is approximately 85%. Falsely positive elevated serum levels of chromogranin A may occur when patients take a commonly prescribed proton pump inhibitor (PPI) or H2 receptor antagonist, so these medications should be discontinued for several days prior to testing and testing should be done fasting. Urine 5-hydroxyindoleacetic acid (5-HIAA), the primary metabolite of serotonin, is elevated in carcinoid syndrome. Test characteristics of this assay are sensitivity of 35% and specificity of 100%. Abnormal levels (>5 mg/24 hours) of 5-HIAA are diagnostic of carcinoid syndrome. Elevated levels of serum serotonin are also confirmatory and consistent with carcinoid syndrome, though more difficult to measure reproducibly. Patients need to be on a special diet in order to avoid false positive results. Bananas, pineapple, tomatoes, plums, eggplant, avocado, kiwi, fruits, and nuts must be avoided for 3 days. Medications such as PPIs, cough and antihistamine medications, thorazine and compazine, nasal drops and sprays, hypertension medications, acetaminophen, and muscle relaxants especially robaxin, valium, and flexeril can also lead to false positives and should be avoided.
Less common NETs may also be identified by the specific hormone and syndrome that is produced. For example, thymic carcinoid tumors may produce adrenocorticotropic hormone (ACTH) and corticotropin-releasing factor and result in ectopic Cushing syndrome.5 Thirty percent of these tumors can also secrete catecholamines; therefore, 24-hour urinary catecholamine excretion should be measured as clinically indicated.
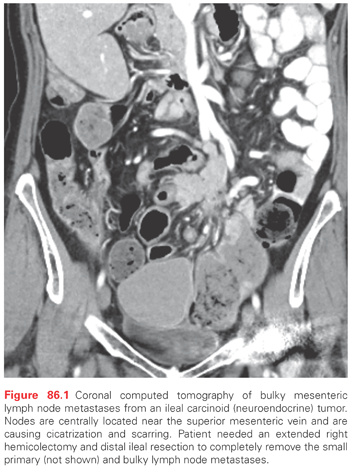
The diagnostic evaluation for GI and pulmonary NETs often begins with high-resolution cross-sectional imaging with either multiphasic computed tomography (CT) or magnetic resonance imaging (MRI). Small intestine NETs are often characterized by mesenteric masses that represent abnormal lymph nodes associated with cicatrization or desmoplastic changes and scarring (Fig. 86.1). Additionally, most NETs express somatostatin receptors that can bind and internalize the currently available octapeptide somatostatin analogues octreotide and lanreotide. This feature can be exploited both in terms of somatostatin receptor-based treatments and scintrigraphy (SRS). 111In-pentetreotide scintigraphy (Octreoscan; Mallinckrodt, Dublin, Ireland) can image approximately 80% to 90% of patients and can unmask the primary tumor, regional lymph node metastases, and distant metastases. Positron emission tomography (PET) scanning with Ga-68 DOTATATE is a newer somatostatin-based scintrigraphy that is thought to be more sensitive than 111In scans. At present, this imaging modality is available primary in Europe and select centers in the United States such that it is still considered experimental. Standard PET scanning with 18F-deoxyglucose is disappointing in patients with well-differentiated GI and pulmonary NETs, although poorly differentiated NETs are often 18F-deoxyglucose avid. 123I- or 131I-meta-iodobenzylguanidine is taken up and accumulated in some NET cells. Sensitivity of 131I-meta-iodobenzylguanidine scan in patients with small intestine NETs is between 60% and 85%, and is especially useful in patients whose tumors secrete catecholamines. CT-enteroclysis is a newer imaging modality designed to image tumors of the small bowel. It has a sensitivity of 86% and a specificity of 100% for small bowel NETs.13 Echocardiography is also included in the diagnostic workup to either confirm or exclude carcinoid heart disease.
General Staging Principles
The seventh edition of the American Joint Committee on Cancer (AJCC) Staging Manual is the first edition to include specific staging for GI NETs such as stomach, small bowel, appendix, colon, and rectum, and parallels staging for adenocarcinomas of those same sites.14 Multiple papers have been published recently which both validate this AJCC staging and propose some additional modifications to enhance future AJCC versions15,16 (see Table 86.1).
General Management Principles
Management of NETs falls under two main categories: therapies directed at tumor control and therapies directed at controlling symptoms of hormone excess. Additionally, therapies directed at tumor control are further divided based on biologic differences of well- versus poorly differentiated NETs. Unless noted, antitumor therapies will all be for well-differentiated tumors. Carcinoid syndrome and medical management of hormone-related symptoms will be addressed in detail later in this chapter.
Therapies directed at tumor control include surgery. Surgical resection remains the mainstay of treatment for resectable NETs of the GI tract and lungs. The primary goal of surgical intervention is to prolong survival. Yet, in addition to prolonged survival, surgery can also palliate symptoms of obstruction, diarrhea, flushing, and/or pain with eating.17 In the setting of unresectable disease, antitumor therapy is only indicated in the setting of symptoms, tumor bulk, and/or disease progression. In newly diagnosed asymptomatic patients with unresectable or metastatic disease, it is often reasonable to monitor closely without any active treatment.
DIAGNOSIS, STAGING, AND MANAGEMENT BY PRIMARY TUMOR SITE
The previously mentioned general principles apply to most NETs of the GI tract and lungs. Following are some unique aspects of the diagnosis, staging, and management by primary tumor site. Management here focuses on surgical management of localized disease and management of hormone symptoms. Management of metastatic disease will be addressed separately.
Thymic Neuroendocrine Tumors
NETs of the thymus are rare. Thymic NETs make up between 2% and 7% of anterior mediastinal masses. CT or MRI is commonly used for imaging and the diagnosis. Patients may present with severe Cushing syndrome secondary to ectopic ACTH production.18 Approximately 25% of patients with thymic carcinoids will have MEN-1. Patients typically present with either stage 1 well-differentiated neuroendocrine carcinoma previously termed typical carcinoid syndrome, or stage 2 moderately differentiated neuroendocrine carcinoma previously called atypical carcinoid tumor or stage 3 poorly differentiated or small cell neuroendocrine carcinoma. Surgery (radical thymectomy) is the primary treatment for stage 1 and 2 thymic NETs, whereas chemotherapy is best for stage 3. Five- and ten-year survival for resected thymic NETs is between 20% and 30%. Radiation may help with local control following incomplete resection.
Bronchial Neuroendocrine Tumors
Bronchial NETs appear histologically like intestinal NETs and are not related to cigarette smoking. These tumors are more common in patients with MEN-1. Poor prognostic factors include higher mitotic index, nuclear pleomorphism, vascular and lymphatic invasion, and poorly differentiated growth pattern. The bronchus is the site of a primary NET in approximately 2% of cases. These tumors occur close to the hilum on CT and MRI scan and may be confused with blood vessels. Bronchial NETs are divided into three categories: benign or low-grade malignant, which is the typical carcinoid form; low-grade malignant, which is the atypical carcinoid form; and poorly differentiated, either large cell or small cell. The grading system differs slightly when compared to GI NET grading. The three different categories of bronchial carcinoid tumors have different prognosis from excellent for typical well-differentiated carcinoids to poor for small cell neuroendocrine carcinomas. The well-differentiated tumors are surgically resected with a lobectomy while the small cell tumors are treated primarily with chemotherapy. SRS can be used to complement cross-sectional imaging with multiphasic CT or MRI. Atypical carcinoid tumors have more uptake of deoxyglucose on PET scan, while typical carcinoid tumors have more uptake of sandostatin (octreotide).19 On bronchoscopy, NETs appear as a cherry red mass within the bronchus protruding into the lumen. It is recommended not to biopsy them as they can bleed excessively with biopsy. Bronchial carcinoids are the most common cause of ectopic ACTH syndrome, and Cushing syndrome is severe with usual clinical signs and symptoms plus severe weakness secondary to hypokalemia.20
Esophageal Neuroendocrine Tumors
Esophageal NETs are very uncommon (<1% of GI NETs). They occur predominantly in men at an age >60 years. Symptoms are nonspecific and vary from asymptomatic to indigestion and burning pain. Most tumors are seen in the distal esophagus just proximal to the esophageal-gastric junction. They are seen on endoscopy as a submucosal mass, and they usually dimple the mucosa. Endoscopic ultrasonography and SRS are indicated for staging because lymph node metastases occur in 50% of patients. Endoscopic mucosal resection (EMR) is done if there is no evidence of lymph node metastases and the tumor is amenable to complete excision. Open resection is done for patients with lymph node metastases. The operative procedure is most commonly an Ivor Lewis esophagogastrectomy that requires an upper midline incision and a right thoracotomy. Survival is dependent on stage of disease and adequacy of resection.
Gastric Neuroendocrine Tumors
Gastric NETs are rare tumors and constitute <1% of gastric tumors and 9% of GI NETs (Table 86.2). These tumors arise from the enterochromaffin-like (ECL) cells of the stomach that occur in the gastric fundus and body. Gastric NETs are immunoreactive to histamine, chromogranin A, and synaptophysin. Gastric NETs have three different forms: type 1, type 2, and type 3. Type 1 gastric carcinoids typically occur in a state of chronic atrophic gastritis that results in achlorhydria and hypergastrinemia.21 They occur in 80% of gastric NETs. These tumors are typically multicentric consisting of multiple small gastric polyps and invariably develop from ECL cell hyperplasia and become small polypoid tumors.22 Tumors vary in size from a few millimeters to 1.5 cm. These tumors are almost always benign with minimal risk of invasion or metastases. These are usually treated with repeat endoscopic examination, excision, and EMR of larger tumors. There is a very low risk of lymph node metastases (5%) or distant metastases (2%). Larger tumors (approximately >2 cm) require resection, either endoscopic or open surgery. If gastric surgery is performed, antrectomy is indicated as this will remove the source of the hypergastrinemia and the stimulus for ECL cell growth.
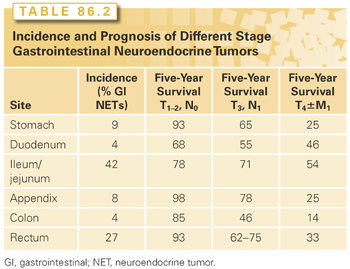
Type 2 gastric carcinoid tumors are rare and occur in only 6% to 8% of patients with gastric NETs. They are much less common than those seen in atrophic gastritis. Type 2 tumors occur in patients with MEN-1 with Zollinger-Ellison syndrome (ZES) who have been treated with PPIs for a long time period. They occur equally in both men and women with an age distribution of 45 to 50 years. The hypergastrinemia of ZES is exacerbated by the prolonged use of the PPI (approximately 10 years) to control symptoms related to excessive acid secretion. These NETs develop in a hyperplasia-dysplasia-neoplasia sequence. Moreover, there must be an effect of the menin gene defect, because non–MEN-1 patients with (sporadic) ZES have also been treated with prolonged use of PPI and have not developed similar tumors. Type 2 NETs of the stomach are often multiple and small (73% <1.5 cm), but are larger than the type 1 tumors. Lymph node metastases are present in 30% of patients with type 2 gastric carcinoid tumors, and distant metastases occur in 10% to 20%. Patients with MEN-1 may develop distant metastases from pancreatic NETs, so it is sometimes difficult to tell which primary NET led to the development of a liver NET. However, rare cases of highly malignant gastric NETs with a poor prognosis have been described in some patients with MEN-1/ZES. These patients have diffuse involvement of the entire stomach with NETs, and total gastrectomy with adjacent lymph node dissection is recommended.
Type 3 gastric NETs are those that occur sporadically with no association of hypergastrinemia. They represent 15% to 20% of gastric NETs and are markedly different from type 1 and 2 NETs. They occur sporadically, are solitary, and grow much more rapidly. Most have distant metastases at the time of diagnosis. These tumors have a male predominance and occur 3:1 in men. Mean age at presentation is 50 years. These tumors are large (mean size 3 cm). Most lesions invade the full thickness wall of the stomach. They are usually located in the body or fundus. Regional lymph node and liver metastases are present in 70% of patients. A total of 73% of patients with this disease are alive at 5 years, but those with liver metastasis have a 5-year survival of 10%. Sporadic type 3 gastric NETs usually have a Ki67 index of >2%. According to older classifications, these tumors may have typical or atypical histology. Atypical implies nuclear pleomorphism, more mitosis, and necrosis. Atypical tumors are larger (>5 cm) and have a more unfavorable prognosis. The atypical carcinoid syndrome occurs in these patients and is associated with tumor release of histamine. The atypical carcinoid syndrome has bright red cutaneous flushing, edema, itching, wheezing, and lacrimation. In patients with type 3 gastric NETs, tumor debulking of the primary and lymph node metastases may relieve symptoms. Hepatic metastases are treated with resection, hepatic artery embolization or chemoembolization, radiofrequency ablation, and sandostatin long-acting repeatable (LAR) that may ameliorate the symptoms of the atypical carcinoid syndrome. Chemotherapy is likely to be useful when the proliferation rate exceeds 5%. Response rates are between 20% to 40%.
Small Bowel Neuroendocrine Tumors
NETs of the duodenum are rare and comprise <4% of all GI NETs. Gastrinomas are the most common functional duodenal NET and represent 60% of duodenal NETs. These tumors are more common in the proximal duodenum and are less likely to arise in the distal duodenum. They are usually small <5 mm and multiple in MEN-1. They frequently (30% to 70%) spread to regional lymph nodes such that adjacent lymph node sampling is recommended but do not commonly spread to distant sites. Overall, the 10-year survival of duodenal NETs is 64%.23 The 10-year survival rate for patients with duodenal gastrin-secreting NETs is 90%. Duodenotomy (opening the duodenum) at the time of surgery is the most effective method to identify these tumors. When found, complete excision of the duodenal wall around the tumor with lymph node sampling is recommended. Some advocate Whipple procedure, but the prognosis is excellent with local tumor resection and the cure rate is 60%, so many think that Whipple resection is not indicated.
Periampullary duodenal NETs do not usually produce a hormonal syndrome.23 Given their location, these tumors may cause obstructive jaundice or bleeding. The size of these tumors is small (between 1 to 2 cm in diameter). They are typically nodular, polypoid, and ulcerated. Periampullary NETs are associated with von Recklinghausen neurofibromatosis. A total of 50% of the patients have either lymph node or liver metastases. They require either local excision or Whipple pancreaticoduodenectomy, dependent on the size of the tumor, the age of the patient, and the relationship to the ampulla. Nonampullary duodenal NETs that are found during endoscopy have an excellent prognosis and can be removed by EMR if <1 cm. They are removed by surgical excision if the tumor is >2 cm. Lymph node metastases can occur in approximately 40% of patients, so lymph node sampling is recommended. For treatment of NETs >3 cm, Whipple pancreaticoduodenectomy is recommended. Small bowel NETs are most common GI NET, and they are most prevalent within the ileum. They account for 42% of all GI NETs. They usually occur within 20 cm of the ileal-cecal valve. They feel like a firm mobile nodule within the wall of the bowel. Patients typically have a long history of vague nonlocalizing abdominal pain before the tumor is detected. These symptoms may be borborygmi, episodic abdominal pain or cramping, and episodic diarrhea and constipation. Others may develop clinical signs of the typical carcinoid syndrome that include diarrhea, flushing, palpitations, intolerance of certain specific foods like cheese or red wine, intestinal venous congestion, and infarction, and these can occur as the tumor lymph node metastases enlarge and block the venous outflow. Intermittent severe episodes of abdominal pain or even intestinal obstruction can occur as the tumor progresses. Ileal NETs are commonly very small (2 to 4 mm) and multiple within the wall of the ileum, with adjacent large lymph node metastases that cause cicatrization and venous conjestion that can result in small bowel obstruction (see Fig. 86.1). Approximately 50% of patients will have liver metastases or peritoneal carcinomatosis at the time of diagnosis. The 10-year survival for jejunal and ileal NETs is 53% and 50%, respectively. Surgery includes wide resection of the small bowel with the primary tumors, its mesentery, and lymph node metastases. This usually requires an extended right hemicolectomy and may result in a relative short gut syndrome because the lymph node metastases can be very centrally located and require resection of proximal branches of the superior mesenteric artery and vein. Recent studies also suggest that surgery can be effectively done laparoscopically,24 but commonly extensive nodal disease is present such that the superior mesenteric artery and vein must be skeletonized that usually requires open surgical techniques. Surgery for ileal NETs is the mainstay of treatment and surgical resection of the primary tumor even in the setting of numerous distant metastases has been recommended in order to avoid the cramping and bowel obstruction associated with primary NETs that are left to progress.25 Patients with unknown primary metastatic NETs can often harbor occult primary ileal NETs. This is quite common, and careful exploration and palpation of the ileum allows detection of small submucosal primaries that feel like little peas within the bowel wall.26 Cholecystectomy is also indicated because most of these patients with either lymph node or liver metastases will require the long-term use of somatostatin analogues that can cause gallstones when administered long-term.
Appendiceal Neuroendocrine Tumors
Appendiceal NETs represent between 5% to 8% of GI NETs. They occur in approximately 1 in 200 to 300 appendectomies.27,28 Most are located in the tip of the appendix. They commonly present in younger patients who have a mean age of <50 years. They seldom metastasize; lymph node metastases occur in 3.8% and distant metastases in only 0.7%. The long-term survival is nearly 95% for all patients with appendiceal carcinoid tumors, 84% for those with lymph node metastases, and 28% for those with liver metastases. Appendectomy is adequate for tumors <2 cm and those that do not invade through the wall of the appendix or are present at the base. For patients with tumors >2 cm, invasion through the appendiceal wall, presence at the base, or lymph node metastases, a right hemicolectomy is indicated.
Colorectal Neuroendocrine Tumors
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


