Cancer stem cells
Yadwinder S. Deol, PhD  Jill Granger, BSc, MSc
Jill Granger, BSc, MSc  Max S. Wicha, MD
Max S. Wicha, MD
Overview
There is substantial evidence that many if not most tumors contain a subpopulation of cells that display stem cell properties. These “cancer stem cells” (CSCs) play an important role in tumor initiation and propagation. Furthermore, these cells may mediate resistance to cancer therapeutic agents and therefore play a fundamental role in cancer relapse. This highlights the importance of developing therapeutic approaches to target this cell population. In this chapter, we will review current thoughts on the role of tissue stem cells in carcinogenesis, the pathways that regulate these cells, and current progress in development of CSC targeting therapeutic agents. We will then review clinical implications of CSC models.
Cancer stem cell hypothesis
The idea that cancer originated from “primitive embryonic-like cells” dates back over a hundred years.1 However, it is only within the past several decades that cellular and molecular technologies have permitted the direct testing of these concepts. The “cancer stem cell (CSCs) hypothesis” consists of two separate but inter-related concepts. The first concerns the cellular origins of cancer and the second the cellular organization of established cancers.
Models of carcinogenesis and cellular origin of cancer
Currently, two main models of carcinogenesis have been proposed, which are summarized in Figure 1. The classical “stochastic” model proposes that cancer may arise from any cell and that carcinogenesis evolves through random “stochastic” processes of mutation followed by clonal selection. As illustrated in the sequential accumulation of mutations during carcinogenesis (Figure 1b), disease development is driven by Darwinian selection of the fittest clones of cancer cells. In contrast to stochastic models, the CSC model posits that this process originates in those cells that possess or acquire the stem cell property of self-renewal. It is important to emphasize that the CSC model does not hold that tissue stem cells are the sole cellular source of cancer initiation. Although cancers may arise in normal tissue stem cells,2, 3 there is evidence that some cancers arise in tissue progenitor cells through mutations that endow these cells with self-renewal capabilities.4 This process generates tumors that display a degree of hierarchical organization. At the apex of this hierarchy are CSCs, which are operationally defined as cancer cells that maintain the ability to self-renew (Figure 1a). These CSCs are capable of generating tumors that recapitulate the phenotypic heterogeneity of the original tumor when transplanted into mouse models.
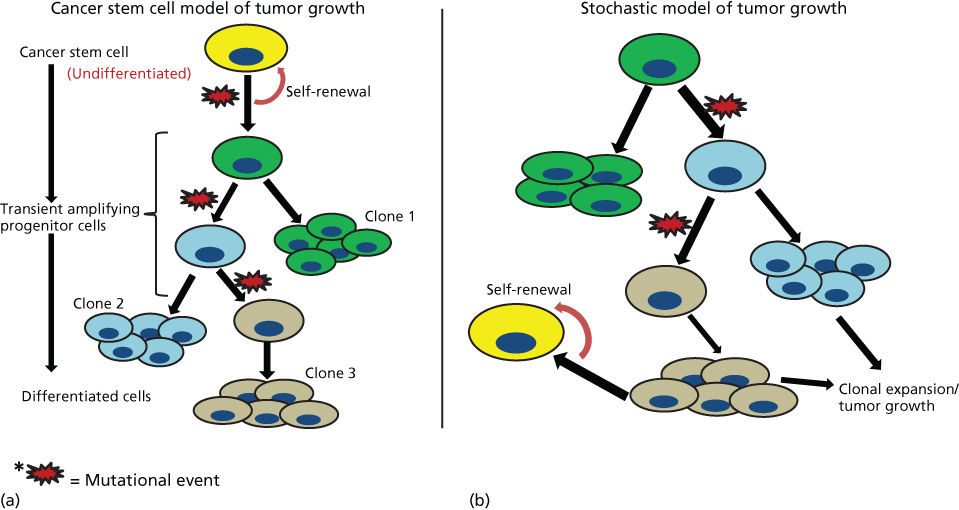
Figure 1 Schematic representation of two models of tumorogenesis. (a) Depicts “cancer stem cell” model where tumors arise in cells with stem cell properties generating hierarchically organized tumors driven by “cancer stem cells” at the top of hierarchy. These cells then generate differentiated clonal progeny that form the tumor bulk. (b) Depicts stochastic or classical model of tumorogenesis where random mutations in any cell generates cancerous clonal populations, which evolve through further mutation.
Although the “stochastic” and “CSC model” were initially described as mutually exclusive, more resent research suggests that elements drawn from both models might better describe tumor development. Recent reports by the Vogelstein suggest that the incidence of cancers in many organs is directly proportional to their number of tissue stem cell divisions.5 The latter is reflective of their stem cell frequency and division rate. As tissue stem cell mutations are themselves stochastic, this work provides a unifying model of carcinogenesis, whereby stem cells may be a functional unit of mutation and subsequent clonal selection. Furthermore, CSCs may undergo mutation during tumor evolution, generating tumors that contain multiple CSCs and their clonal progeny. The combination of these concepts, derived from both models of carcinogenesis, may provide a molecular explanation for the generation of intratumor heterogeneity; a phenomenon with considerable therapeutic implications.
Cancer stem cells as a unifying concept in multiple cancers
CSCs were first identified in human leukemia in 1997.6 In seminal studies, John Dick et al. demonstrated that only a small fraction of primary human leukemia cells were capable of regenerating the leukemia when transplanted into immune-suppressed NOD/SCID (nonobese diabetic/severely compromised immunodeficient) mice.6 Further, these tumor-initiating leukemia cells were prospectively identified by virtue of their phenotypic characterization as CD34+/CD38−, a phenotype resembling that found on normal hematopoietic stem cells. Although the frequency of these leukemia initiating cells was found to be approximately 1 in 250,000, the leukemias generated in transplanted mice recapitulated the phenotypic heterogeneity of the initial tumor. Utilizing similar tumor transplantation technologies, investigators have subsequently identified CSC in a wide variety of solid tumors. These include cancers of the breast,7 brain,8 prostate,9, 10 colon,11, 12 pancreas,13 liver,14, 15 lung,16 melanoma,17 and head and neck.18 In fact, evidence suggests that the majority of tumors are hieratically organized and contain cell populations that display stem cell properties.
Isolation, identification, and characterization of CSCs
In order to study the basic attributes of CSCs, it is necessary to first identify, isolate, purify, and characterize these cells by employing techniques that enable one to distinguish them from the bulk tumor cell population. The functional characteristics of CSCs, including self-renewal capability and differentiation potential, can be exploited by various in vitro and in vivo assays to identify and characterize this important cell population. Several methods or techniques exist to identify and isolate stem cell-like cancer cells; flow cytometry detecting expression of CSC-related markers, dye exclusion, ALDH (aldehyde dehydrogenase) assay, label retention (PKH staining), and a recent novel method of using autofluorescence in presence of riboflavin. CSCs, enriched by these techniques, may be functionally validated by sphere formation assays or serial transplantation assays in vivo for evaluation of tumorigenic and self-renewal potential. The details of the methods are listed in the following sections.
CSC marker expression
The first CSCs in solid tumors were described in human breast cancer based on cellular expression of CD44 and lack of expression of CD24 (i.e., CD44+/CD24−).7 Interestingly, these markers have proven useful for the isolation of CSC from a variety of other solid tumors. In parallel, CD133 has been shown to be useful in enriching for CSC in brain and lung cancers.8, 16 Another stem cell marker, ALDH, is also used along with CD44/CD24 or CD133 to identify the CSC population in several tumors. ALDHs are a family of enzymes that are involved in the oxidation of retinol to retinoic acid.19 ALDH activity can be readily determined using the commercially available Aldefluor assay and was shown to be a useful CSC marker in cancers of the breast,20 ovary,21 colon,22 head and neck,23 and recently melanoma.24
Dye exclusion assays
Dye exclusion is based on the ability of stem cells to efflux lipophilic dyes, such as Rhodamine or Hoechst, owing to the high expression of the ATP-binding cassette transporter proteins such as ABCG2/BCRP1.25 Utilizing flow cytometry analysis, cells that do not retain dye are observed as a side population.26 Several studies have demonstrated that such side populations are enriched in cells with tumor-initiating capacity and are capable of regenerating heterogeneous cell populations.27 This stem cell-like side population has been observed in several cancer types, as well as bone marrow and normal solid tissues.28, 29 The cellular toxicity of Hoechst dye limits their ability to be combined with functional studies.
Label retention
Another dye-based method used to characterize CSCs in vitro is the cell membrane, label-retaining assay using fluorescent PKH dye. This dye consists of a fluorophore that binds to the cell membrane lipid bilayer.30 Upon cell division, the dye becomes distributed equally among daughter cells. Subsequent continuous cell divisions lead to decreased intensity.31 Stem cells, including CSCs, normally undergo an asymmetric self-renewal process generating a stem cell and a daughter cell. The generated stem cells are quiescent and thus retain PKH dye for a longer time period, whereas the daughter cells proliferate and differentiate leading to decreased intensity of PKH dye. These daughter cells are then identified and sorted by flow cytometry. This method has been used successfully to isolate murine hematopoietic and mammary stem cells.32
Recently, it was reported that CSCs demonstrate autofluorescence in the presence of riboflavin.33 The authors isolated this population using flow cytometry, distinguished these cells from the side population, and demonstrated that these putative CSCs had a greater tumor sphere-forming ability, increased chemo-resistance, were highly metastatic, and possessed long-term in vivo tumorogenecity. Further, in support of the cancer stem model, this autofluorescent CSC-like population was detected in several cancers.33
Sphere formation assays
The sphere formation assay is another in vitro assay that has proven useful for the identification of CSC populations. This is based on a property shared by both normal and malignant stem cells: survival when cultured in suspension and generation of spheroids at clonal density.34 These spheres can be serially passaged and display stem cell properties with each passage. It has also been possible to combine a number of in vitro CSC assays. For instance, the addition of PHK dye during sphere formation results in the formation of spheres that often contain a single label-retaining sphere-initiating cell.35
In vivo serial dilution CSC assays
Although the use of CSC markers and in vitro assays has proven useful in the isolation and characterization of CSC, all of these techniques have limitations. The expression of CSC markers is variable and may be influenced by culture conditions or the tumor microenvironment.36 Further, sphere formation assays may not correlate with tumor initiation capacity.37 In light of these limitations, the “gold standard” for CSC identification is demonstration of tumor initiation upon implantation into immunosuppressed mice.37 The tumor-initiating frequency in any cell population may be assessed by the ability of cells to initiate tumor development in mice using limiting dilution of cell suspensions. Thus, the definition of “CSCs” is ultimately an operational one.
It is important to note that immunosuppressed mouse models also have limitations. In human melanoma, the frequency of tumor-initiating cells was strongly influenced by the degree of immune competence of the mouse model utilized.38 Recent studies have indicated that the immune system may actually play an important role in CSC regulation.39, 40 Studies elucidating these roles have been carried out in mouse tumors transplanted into syngeneic immune-competent mice. Such studies have confirmed the existence of CSC as determined by serial transplantation.41
Lineage tracing studies
One criticism of all transplantation studies is that they perturb the normal microenvironment where tumors develop. To circumvent this limitation, a series of studies using cell markers and lineage tracing have been performed. Such studies have confirmed the existence of CSC in tumors of the skin,42 brain,43 and colon.44
EMT/MET and cancer stem cells
Epithelial–mesenchymal transition (EMT) is a physiological process of particular importance during embryonic development whereby epithelial cells acquire mesenchymal properties, are characterized by a loss of cell adhesion and the acquisition of invasive properties. The EMT process was initially described during development, where it induced the migration of mesodermal cells during gastrulation or the delamination of the neural crest cells from the dorsal neural tube.45 Similarly in cancer, EMT has been associated with the process of metastasis where epithelial cells from the primary tumor acquire mesenchymal phenotype and invade to other tissues.46
EMT is regulated by signals from the microenvironment that mediate epigenetic alterations driven by core transcription factors. Microenvironmental factors include tissue hypoxia, TGF-β (transforming growth factor-β), and other inflammatory molecules with subsequent induction of EMT-related transcription factor expression such as TWIST1, TWIST2, SNAI1, SNAI2, ZEB1, and ZEB2, among others.47, 48 Overexpression of these transcription factors induce a mesenchymal and invasive phenotype through repression of cell adhesion molecules, such as E-cadherin.49 Twist1, which is involved in mesenchymal development during embryogenesis, has been shown to induce metastasis in breast cancer.50 Several studies suggest that the EMT process generates CSC-like cells.51, 52 In this regard, overexpression of SNAI1 or TWIST1 alone was shown to induce EMT in MCF10A and immortalized human mammary epithelial cells.53 In addition, there was increased expression of the stem cell marker CD44 and a decrease in CD24 expression, which suggests induction of CSC-like phenotype. Moreover, TWIST1 has been associated with an increased ability of breast cancer cells to form mammospheres and secondary tumors in an oncogenic HRAS-g12V model.54 Based on the molecular classification of breast cancer, a basal subtype, particularly claudin-low are known to possess an EMT-like gene expression signature. Such cancers have a high proportion of CD44+/24− cells that are locked within the mesenchymal state. These tumors display a highly aggressive behavior accompanied by a greater propensity to develop metastasis. Further, TWIST1 upregulated BMI expression, when suppressed by the miR-200 microRNA cluster, decreased CSC self-renewal, and cellular differentiation. Similarly, BMP signaling induced MET via induction of miR-205 and the miR-200 family of microRNAs that are key regulators of MET.55
The aforementioned studies strongly suggest the role of EMT in the acquisition of a stem cell phenotype. It is plausible that the CSC possesses alternative phenotypes; one involved in tumor invasion and metastasis while another state maintains the bulk of the tumors. This is evident from the recent study that used gene expression analysis to compare ALDH+ and CD44+/24− BCSCs isolated from different subtypes of breast cancer. This comparative gene expression analysis suggested that, although each population was independently capable of tumor initiation in NOD/SCID mice, yet they were quite distinct both functionally and anatomically in the original primary tumors. ALDH+ CSCs usually populated the interior of the tumors where they self-renewed and proliferated at a higher rate, whereas CD44+/24− CSCs were more mesenchymal, displayed an EMT phenotype located on the tumor margins or invasive front, and were more quiescent.56 Further characterization of these two populations showed that CD44+/24− cells were highly vimentin positive and E-cadherin negative, whereas an ALDH+ population was highly positive for E-cadherin and negative for vimentin. Therefore, these CD44+/24− cells were more EMT-like, and may be poised to invade the blood vessels when triggered by stromal factors such as hypoxia, TGF-β, and other inflammatory agents.
These studies also demonstrated that BCSCs maintain the plasticity to transition between EMT and MET states in a process regulated by the tumor microenvironment. The interconversion of BCSCs from the EMT-like to MET-like state has also been shown to be regulated by microRNA networks. Such networks included EMT-inducing mir-9, mir-100, mir-221, and mir-155 as well as MET-inducing mir-200, mir-205, and mir-93.57 The plasticity of BCSCs from a quiescent mesenchymal state to a proliferative epithelial-like state is critical for formation of tumor metastases at distant sites. The knockdown of TWIST1 in a spontaneous squamous-cell carcinoma model reversed EMT and allowed cells to proliferate and form metastasis.58 Similarly, the loss of the EMT inducer Prrx1 was required for cancer cells to form lung macrometastasis.59 In addition, MET-promoting factors, such as miR-200 family members, were found to promote metastatic colonization of breast cancer cells and induce epithelial differentiation.60 Furthermore, expression of Id1 gene in breast cancer cells reconverted EMT cells into MET by downregulating TWIST1, which is required for establishing macrometastasis.61 The above studies provide evidence that two alternative states of CSCs display plasticity and are regulated by microenvironmental factors that modulate the genetic regulators and several transcription factors. The existence of multiple CSC states has important therapeutic implications as elimination of CSC may require successful targeting of multiple CSC populations.
Signal transduction pathways in cancer stem cells and their therapeutic targeting
Like their normal counterparts, a number of evolutionarily conserved developmental pathways play a significant role in the regulation of CSCs in several cancer types. The conservation of these pathways in CSCs of different tumor types further strengthens the cancer stem model. Moreover, these pathways could provide common potential therapeutic targets irrespective of the tumor type.
Hedgehog signaling
The Hedgehog (Hh) family of proteins controls cell growth, survival, pattern of the vertebrate body, and stem/progenitor cell maintenance. Dysregulation of Hh signaling, including that arising from somatic mutation, has been linked to several cancer types. The binding of Hh proteins to the receptor Patched (Ptc) activates a signaling cascade that leads to the upregulation of a zinc-finger transcription factor GLI1–3 and its downstream gene targets.62–64 Of the three mammalian Hh ligands, sonic hedgehog (SHH), Indian hedgehog (IHH), and desert hedgehog (DHH), SHH is the most highly expressed Hh signaling molecule and is involved in the regulation of many epithelial tissues. Aberrant activation of Hh signaling has been linked to basal cell carcinoma,65, 66 medulloblastoma,67, 68 rhabdomyosarcoma,69 and other cancers, such as glioma, breast, esophageal, gastric, pancreatic, prostate, and small-cell lung carcinoma.70 GLI1 amplifications are also known to occur in gliomas.71
The Hh signaling pathway plays a major role in mammary gland development both in humans and in mice. In mice, constitutive activation of Gli1 or inactivation of Gli3 resulted in mammary bud formation defects.72 Ectopic expression of Gli1 has been shown to induce the nuclear Snail expression resulting in loss of E-cadherin expression in mouse mammary glands tissue during pregnancy.73 In addition, the transcription factor FOXC2 also promoted mesenchymal differentiation via the Hh pathway and Snail upregulation.74 Similarly, SHH overexpression in mouse mammary glands leads to mammary bud defects.75 Perturbation of other Hh pathway molecules, such as activation of SMO or loss of PTCH1, has resulted in terminal end bud abnormalities.76 Normal human mammary stem cells expressed PTCH1, Gli1, and Gli2 during mammosphere formation, a pathway that is downregulated during the process of differentiation. Hh signaling played an important role in stem cell regulation as activation of Hh signaling through Hh ligand, or Gli1/Gli2 overexpression, increased mammosphere formation and size through activation of BMI-1, a member of the polycomb gene family.77 The similar activation of SMO, driven by an MMTV (mouse mammary tumour virus) promoter, increased mammosphere formation of primary mammary epithelial cells in transgenic mice.76 Hh ligands are also known to exert mitogenic effects on mammary stem cells through paracrine interactions. This process may be involved in the regulation of mammary differentiation during pregnancy, in a process regulated by TP63.78 Apart from TP63, the transcription factor Runx2 has been shown to regulate the expression of Hh ligands.79
Hh signaling pathway members SHH, PTCH1, and GLI1 were highly expressed in invasive breast carcinomas suggesting a role for this pathway in human breast cancers.80 Further, Hh ligand expression was associated with basal-like breast cancer; an aggressive phenotype with relatively poor outcome. These findings were corroborated in mouse models where ectopic expression of Hh ligand in BLBC mice induced high-grade invasive tumors.81 Similarly, SHH upregulation due to hypomethylation of the SHH promotor has been observed in breast carcinomas that were associated with expression of NF-κB in breast cancer clinical samples.82 Hh signaling has also been linked to bone metastasis in this disease. In bone metastases, secretion of Hh ligand by tumor cells activates the transcription of osteopontin (OPN) by bone osteoclasts. This further promotes osteoclast maturation and resorptive activity, facilitating metastasis.83
The most commonly used antagonist of the Hh pathway is the plant alkaloid cyclopamine that binds to SMO and downregulates Gli1.84 Other antagonists that bind to SMO include SANTs 1–4;85 KAAD-cyclopamine,84 compound-5 and compound-Z,86 and Cur-61414.87 Further, 5E1 monoclonal antibody targeting SHH treated tumors in small-cell lung carcinoma.88 GDC-0449 (Vismodegib, trade name: Erivedge®), the first Hh pathway inhibitor approved by FDA, was used in clinical trials in combination with the Notch signaling inhibitor RO4929097 (a γ-secretase inhibitor, GSI).89
Notch signaling
Notch is another evolutionarily conserved signaling pathway that is involved in the regulation of cell fate determination, proliferation, and differentiation in many tissues. The core components of the Notch signaling cascade include the ligands-Delta-like-1, -3, and -4 (DLL1, DLL3, and DLL4), Jagged-1 and -2 (JAG1 and JAG2), and four transmembrane receptors (Notch 1 to Notch 4). After ligand binding, the receptor undergoes two proteolytic events. First, cleavage occurs in close proximity to the extracellular side of the plasma membrane. This is followed by a second cleavage within the transmembrane domain, mediated by a γ-secretase complex, which leads to the release of the intracellular domain of the receptor from the membrane. Upon cleavage, the intracellular Notch domain translocates to the nucleus where it forms a complex with the ubiquitously expressed transcription factor CSL and recruits co-activators such as mastermind-like (MAML-1, -2, and -3).90 The transcriptional targets of Notch signaling include not only differentiation-related factors, such as Hairy/enhancer of split (Hes) and Hes-related (HRT/HRP/Hey) families, but also cell cycle regulators (p21 and cyclin D1) and regulators of apoptosis.90, 91
The oncogenic role of Notch 1 was first discovered in T-ALL where the translocation of t(7;9)(q34;q34.3) led to juxtaposition of Notch 1 with TCR-β (T cell receptor-beta). This translocation led to the ligand-independent activity and deregulated expression of intracellular Notch 1 regulated by TCR-β that impacted T-cell differentiation.92 Overexpression of Notch 4 in mouse mammary glands utilizing an MMTV promotor blocked cellular differentiation with aberrant lactation and poorly differentiated mammary and salivary adenocarcinomas occurred between two and seven months of age.93 This evidence again underscores the role that Notch signaling plays in the differentiation and tumorigenesis of several tissues. A correlation between Ras overexpression and elevated Notch 1 levels has been reported.94 Furthermore, expression of Numb, a negative regulator of Notch signaling, was lost in approximately 50% of primary human mammary carcinomas and these tumors showed increased Notch 1 activity.95 Overexpression of several Notch signaling molecules was reported in several tumors such as pancreatic cancer, renal cell carcinoma, prostate cancer, multiple myeloma, and Hodgkin’s and anaplastic lymphoma.96–100 In lung cancer, Notch signaling had a differential effect based on the cell type: in small lung cancer, Notch signaling is inactive but Hes1 and Hash1 are active101 and introduction of constitutively active intracellular Notch induces Notch signaling resulting in growth arrest.102 In contrast, Notch signaling is active in nonsmall lung cancer with high expression of Hes1 and Hey1.101
CD24+/24− breast CSCs has been reported to display Notch pathway activation.103 Notch 3 was found to be upregulated in CD44+ populations of normal and breast cancer cells via SAGE analysis.104 Other studies have suggested that Notch 4 is the most important regulator of breast stem cells.105 Moreover, aberrant activation of Notch signaling is an early event in breast cancer, as this has been observed in ductal carcinoma in situ (DCIS). Treatment of primary DCIS tissue with a γ-secretase inhibitor significantly reduced the mammosphere formation.103 Similarly, in an in vitro mammosphere culture system, activation of Notch signaling via a DSL peptide promoted self-renewal and branching morphogenesis in three-dimensional Matrigel cultures,106 a process that was inhibited by a Notch 4 blocking antibody or a γ-secretase inhibitor. Therefore, activated Notch signaling may maintain the epithelial cells in a proliferative state instead of differentiation leading to carcinoma.
Regulation of Notch pathways has also been attributed to epigenetic regulation through hypermethylation and hypomethylation. Notch ligand DLL1 gene is hypermethylated resulting in decreased NOTCH1 expression.107 Similarly, reduced methylation of NOTCH4 gene promoter in tumors compared to surrounding tissue was observed.108 Overexpression of JAG2 in multiple myeloma cells was correlated with hypomethylation in malignant cells from multiple myeloma patients and cell lines.109 Knockdown of the canonical Notch effector Cbf-1 in mammary stem cells increased stem cell activity, whereas constitutive Notch signaling increased luminal progenitor cells, leading to hyperplasia and tumorigenesis.110 In addition, co-expression of JAG1 and NOTCH1 was associated with poor overall survival.111 In ESA+ CD24-CD44+ BCSCs, activity of Notch 4 and Notch 1 were eightfold and fourfold higher, respectively, compared to the differentiated bulk tumor cells and inhibition of this activity resulted in reduce tumor growth.105
The complexity of the Notch pathway increases through interaction with other oncogenic pathways such as ErbB2, Jak/Stat, TGF-β, NF-κB, Wnt, and Hh.112 As ErbB2 induces Notch 1 activity through Cyclin D1,113 combined treatment of DAPT and Lapatinib targeted stem/progenitor cells both in vitro and in vivo in DCIS.114 The Wnt pathway also interacts with Notch through Jagged-1 (target of WNT/TCF pathway) and Mel-18, a negative regulator of Bmi-1. Knockdown of Mel-18 has been shown to enhance the self-renewal of BCSCs through upregulation of Jagged-1, consequently activating the Notch pathway.115 The activation of Notch activity further activates the Hh pathway and increases expression of Ptch and Gli.77 Taken together, these studies suggest that treatments aimed at targets affecting multiple stem cell pathways could present a novel strategy for targeted therapies. A variety of agents that inhibit Notch signaling are in early phase clinical trials. These include γ-secretase inhibitors that block Notch processing,116 antibodies against specific Notch receptors,117, 118 and antibodies against the Notch ligand DLL4.119, 120
Wnt signaling
The Wnt/β-catenin/TCF pathway is another complex evolutionarily conserved pathway involved not only in development and maintenance of adult tissue homeostasis but also cell proliferation, differentiation, migration, and apoptosis.121 It also regulates the self-renewal and maintenance of embryonic and tissue-specific stem cells.122–124 Dysregulation of Wnt signaling is a hallmark of several cancers where molecules involved in pathway could act as either tumor suppressors or enhancers depending on their activation status.125–127 Wnt signaling involves both canonical and noncanonical pathways. The canonical Wnt pathway involves activation of β-catenin/TCF complex, resulting in its dissociation from APC (adenomatous polyposis coli) with subsequent translocation into the nucleus leading to activation of transcriptions including TCF/LEF.128 This in turn leads to upregulation of oncogenic targets such as c-myc and cyclin D1. This canonical pathway has been extensively studied and found to be deregulated in many cancers. Noncanonical Wnt signaling involves a planer-cell polarity pathway that regulates the cytoskeleton and a Wnt-calcium-dependent pathway that regulates intracellular calcium levels.129, 130 Mutation of APC (Wnt pathway molecule regulating β-catenin stability) is associated with the majority of sporadic colorectal cancers.131, 132 Germline mutations in APC have been shown to be involved in familial polyposis syndromes, a condition associated with development of numerous intestinal polyps with a high propensity for further neoplastic transformation. In addition to colon cancer, oncogenic mutations in β-catenin have been described in cancers of the liver and colon, as well as in melanoma, thyroid, and ovarian cancers.133 Epigenetic silencing through methylation of Wnt antagonist genes such as the secreted frizzled-related proteins (SFRPs) has been reported in colon, breast, prostate, lung and other cancers.121
Wnt signaling has been shown to be involved in maintenance of CSCs in several cancers. In a transgenic mouse model, LRP5 (a key component of the Wnt co-receptor group) knockdown significantly reduced stem/progenitor cells.134 In addition, transgenic mice overexpressing Wnt-1 developed mammary glands that were enriched for epithelial cells expressing keratin 6 and Sca1 markers of mammary stem cells. This suggests that mammary carcinogenesis may involve stem cell expansion secondary to Wnt activation. Wnt/β-catenin signaling is also involved in EMT regulation through downregulation of E-cadherin and upregulation of Snail and Twist.135 EMT has been shown to induce stem cell-like phenotype in breast cancer cells.52 Wnt signaling has also been demonstrated to maintain pluripotency and neural differentiation of embryonic stem cells.136 Wnt signaling may also contribute to stem cell expansion induced by tumor hypoxia, a process mediated by Hif 1-α.137
A number of drugs that inhibit Wnt signaling are in development. In preclinical models, two nonsteroidal anti-inflammatory drugs (NSAIDs), sulindac138 and celecoxib,139, 140 have been found to inhibit Wnt signaling by targeting Dvl and cyclo-oxygenase, respectively. Sulindac is currently being investigated in phase II trials.141 In addition, several non-NSAID inhibitors such as NSC668036 and PCN-N3, among others, have been shown to degrade β-catenin by inhibiting DVL and hence, stabilizing the destruction complex.142, 143 Further, antibodies targeting Wnt pathway members are also under development. A Wnt3A neutralizing antibody was shown to reduce proliferation and enhance apoptosis in prostate cancer mouse models.144 Another monoclonal antibody-OMP-54F28 has been shown to inhibit patient-derived xenograft tumor growth by specifically targeting CSCs. A Wnt decoy receptor145 and an antibody to the Wnt co-regulator R spondin122 are also in development. These aforementioned Wnt inhibiting drugs are currently being tested in several early phase clinical trials.
Clinical significance of cancer stem cells and future perspectives
There is now substantial evidence that CSC plays an important role in treatment resistance; in addition to their role in tumor propagation and tumor metastasis, resistance of CSC to cytotoxic chemotherapy was first demonstrated in preclinical models146, 147 and later confirmed in clinical trials.148 The relative resistance of CSC to radiation therapy has been demonstrated in a number of tumor types. In patients, it has been shown that the relative proportion of cells expressing CSC markers increases following chemotherapy or radiation therapy.148 A number of mechanisms have been described that contribute to this resistance. These include alterations in cell cycle kinetics, increased expression of anti-apoptotic proteins, increased in cellular transporters, and increased efficiency of DNA repair, as reviewed in Ref. 149. In addition to the relative therapeutic resistance of CSC, these cells may actually be stimulated by chemotherapy via stimulation of cytokine loops activated by these treatments. These inflammatory cytokines include IL-8 and IL-6. Inhibitors to these cytokines and their receptors have been developed, and several are entering clinical trials.
The CSC model has significant clinical implications. In addition to emphasizing the importance of successfully targeting these cells, acknowledgement of their existence implies a critical role in the tumor microenvironment, which merits thoughtful consideration for the future design of therapeutic trials and development of appropriate clinical endpoints. Currently, tumor shrinkage as accessed by RECIST (response evaluation criteria in solid tumors) is considered an important endpoint for accessing treatment efficacy. However, tumor shrinkage is only poorly correlated with ultimate patient survival across a wide spectrum of tumor types.150 Tumor shrinkage is a measure of effects of treatments on bulk tumor populations. As CSC constitute only a minority of the tumor bulk, successful targeting of these cells will not necessarily lead to tumor shrinkage. For that reason, the current approach for CSC drug development involves accessing toxicity in Phase I trials followed by combining CSC-targeting drugs with more traditional agents that target bulk populations. As many of the pathways utilized by CSC are shared with normal tissue stem cells, caution is required when introducing such therapeutics in the clinic to access their safety profile. With this cautionary note, a number of CSC-targeting drugs summarized in Figure 2 are now in early phase clinical testing. Interestingly, preliminary data suggests that these agents are well tolerated at doses that reduce CSC number in serial biopsies.151 This suggests that CSC may be more dependent on these pathways than their normal tissue stem cells counterparts, providing an advantageous therapeutic window. Novel methods are under development to achieve more direct measurement of CSC targeting agents’ effects. These include accessing CSC by marker expression in circulating tumor cells (CTC) assays. Studies have demonstrated that CTCs are highly enriched for CSCs supporting the important role these cells play in mediating tumor metastasis.152–154
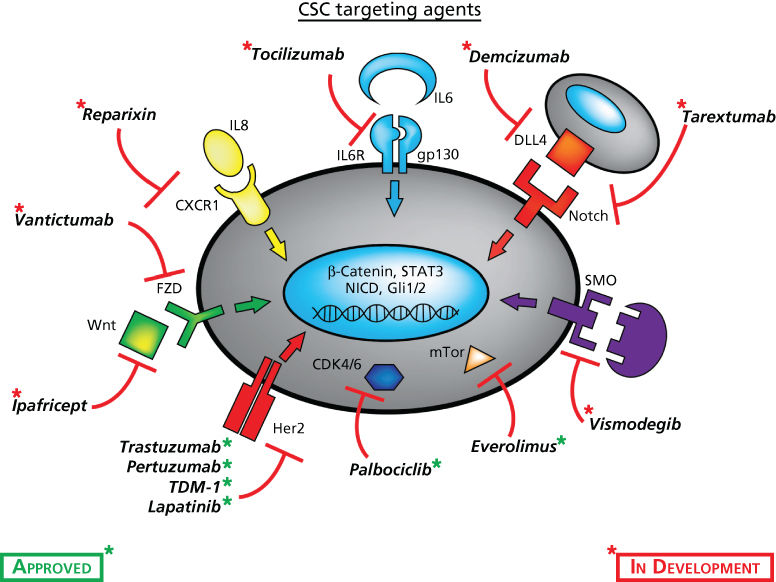
Figure 2 Cancer stem cell-targeted drugs and their cellular targets currently being evaluated in early phase clinical trials or FDA approved for other purposes.
Another important prediction of the CSC model is that CSC-targeted therapies should have their greatest effect when they are deployed in the adjuvant setting. This is the case because the CSCs are unique in that they possess a sufficient self-renewal capacity to generate clinically significant disease from micrometastases. The efficacy of adjuvant therapies is directly related to their ability to eliminate micrometastasis. This concept is supported by studies demonstrating the remarkable ability of Her 2-targeting drugs, such as trastuzumab, to prevent recurrence when utilized in the adjuvant setting in Her2+ breast cancer.148 The clinical importance of Her2 in breast cancer may be due to its role as an important regulator of CSC in these tumors.155
One of the most exciting areas in cancer therapy is the development of cancer immunotherapies including immune checkpoint blockers. Interestingly, there is evidence that CSC may be particularly competent at evading immune surveillance. Several mechanisms have been proposed, including the high expression of PDL-1 or secretion of immunosuppressive TGF-β. A number of approaches utilizing the immune system to target CSCs are being developed to circumvent these processes. These include CSC-based vaccines and peptides. In the future, such approaches may be combined with immune checkpoint blockers to more effectively target CSCs.
Worldwide, there are currently over 70 clinical trials utilizing novel CSC targeting agents. Most of these studies are in their early stages but the next several years should yield exciting and potentially significant results. Ultimately, carefully controlled randomized clinical trials will be required to conclusively determine if the successful targeting of CSCs improves patient outcome.
Summary
Despite considerable progress in delineating the molecular underpinnings of cancer, this increased understanding has yet to translate into significant improvements in survival for patients with advanced disease. For most common cancers, the development of metastasis renders them incurable. There is now substantial evidence that many cancers are hierarchically organized and driven by a population of cells that display stem cell properties. These properties include the ability to self-renew and differentiate, forming the cells that constitute the tumor bulk. These cells, termed “tumor-initiating cells” or “cancer stem cells,” may mediate tumor metastasis and contribute to treatment resistance. The existence of as well as understanding of molecular mechanisms regulating cancer stem cells is of clinical significance in that additional therapies targeting stem cell populations may be required to limit metastasis and significantly improve patient survival.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


