Cancer metabolism
Natalya N. Pavlova, PhD  Craig B. Thompson, MD
Craig B. Thompson, MD
Overview
Successful cell proliferation is dependent on a profound remodeling of cellular metabolism, required to support the biosynthetic needs of a growing cell. Constitutive signaling through aberrantly activated oncogenes activates pro-growth metabolic circuits that can lock cancer cells into net macromolecular synthesis and resistance to cell death. Oncogenic signaling can induce cellular uptake of metabolic substrates, such as glucose and glutamine, as well as utilization of unconventional nutrient sources. In contrast to nonproliferating tissues, which preferentially oxidize acquired metabolic substrates in mitochondria to fuel the oxidative phosphorylation and energy production, cancer cells prioritize the use of acquired metabolic substrates in anabolic processes, including fatty acid, cholesterol, nonessential amino acid, and nucleotide biosynthesis. The accumulation of these precursors supports the synthesis of the macromolecules necessary to sustain cell growth. Oncogene-directed increases in levels of select metabolites can also influence cellular processes beyond metabolic circuits, resulting in changes to the cellular epigenetic state as well as long-ranging effects on cells within the tumor microenvironment. Recent studies suggest these alterations in tumor cell metabolism can be exploited to improve the diagnosis and treatment of a wide variety of cancers.
Malicious builders
A unifying characteristic of cancer is loss of extrinsic controls over cell proliferation. In normal tissues, the quantities of cells, as well as their relative positions, are maintained by extracellular signaling, which include tissue-specific soluble growth factors as well as signals from the extracellular matrix to which cells adhere. Together, these mechanisms control the timing and the extent of cell survival and proliferation. Basal level of signaling enables day-to-day tissue maintenance, while the upregulation of growth signaling in response to tissue perturbations leads to enhanced production of terminally differentiated cells—for instance, epidermal keratinocytes and dermal fibroblasts in a setting of a cutaneous injury, or T cells in response to infection. In this manner, cell proliferation only takes place in response to a cell-type-specific signal of a sufficient strength and duration, which ensures that the overall structural and biochemical integrity of a metazoan organism is maintained.
In contrast to normal cells, genetic and epigenetic alterations acquired by cancer cells allow them to accumulate within tissues in a manner that is independent of extrinsic growth stimuli, at the same time becoming resistant to antigrowth signals.1 Together, these adaptations enable survival and proliferation of cancer cells in settings where normal cell proliferation is strictly regimented. Mutations, copy number amplifications, and translocations targeting growth factor receptors are among the most frequently seen growth-promoting genetic alterations in tumors and are thus termed “driver alterations.” For example, activating mutations in EGFR (epidermal growth factor receptor) are found in 10% of nonsmall cell lung carcinoma in United States and up to 35% in Japan,2 and amplifications of HER2 are found in 25–30% of breast carcinomas.3 In addition to growth factor receptors, downstream integrators of growth factor and matrix attachment signals are often aberrantly activated in cancer as well: for instance, up to 70% of breast cancers have mutations in the PI-3 kinase pathway,4 up to 90% of pancreatic tumors are driven by activating mutations in KRAS,5 and amplification of c-Myc is seen in up to 50% of human cancers regardless of tissue of origin.6
A cell that had received a proper growth signal commits to creating two daughter cells. To accomplish this, it must synthesize a complete set of biomolecules sufficient to build a new, functional cell. This set of biomolecules is extremely diverse and includes not only a vast array of proteins, but also lipids and their derivatives for building plasma and organelle membranes, hexose sugars for glycosylation of proteins, as well as pentose sugars and nucleotide bases for genome replication. To ensure that a cell does not attempt to replicate itself without having ample supply of biomolecules, the same signals that control proliferation are in control of nutrient uptake and the biosynthesis of macromolecular precursors. Thus, a growth-factor-stimulated cell undergoes remodeling of its metabolism in order to support the required biosynthesis. First, this remodeling involves acquiring a sufficient amount of building materials from the surrounding environment. Second, a proliferating cell must prioritize the use of these materials for biosynthesis, as opposed to catabolizing them for energy.
Key Points
Constitutive signaling via aberrantly activated oncogenes allows cancer cells to perpetually receive pro-growth stimuli, which for normal tissues are only transient in nature. As a consequence, cancer metabolism is constitutively geared toward supporting proliferation.
This chapter will describe the mechanisms of how oncogenic transformation reshapes cellular metabolism toward nutrient acquisition and biosynthesis. Furthermore, this chapter will demonstrate how altered metabolic state of cancer cells, in turn, exerts profound effects on cellular epigenetic state and on tumor microenvironment, driving tumor expansion and dissemination. To this end, we will explore how alterations in tumor metabolism can be exploited in cancer detection and therapy.
From yeast to mammals—same means to different ends
Metabolic pathways and enzymes that drive their progression are remarkably conserved throughout evolution, yet the way the metabolism of metazoan cells is regulated is fundamentally distinct from unicellular organisms. Unicellular organisms, such as bacteria or yeast, use external nutrient abundance as a signal to begin proliferating.7 By attuning their proliferation to the environmental conditions, this strategy allows them to maximize their evolutionary success. For a metazoan organism, on the other hand, an evolutionary goal is not to multiply its cells to no end—which would lead to a physiologic chaos—but to maintain the functionality and structural integrity of its organs and tissues, repair injuries, and fight infections. To serve this goal, metazoans have developed complex and tissue-specific control mechanisms, which regulate cell survival and proliferation, yet allow for rapid induction of proliferation of specific cell populations when needed. Accordingly, the pro-proliferative metabolic state of cancer cells, driven by the loss of tissue-specific control mechanisms, can be best understood by first examining the differences in metabolic regulation in metazoans and in unicellular eukaryotes.
Metabolism of living cells is centered on the acquisition and stepwise biochemical transformation of molecules, which consist of reduced carbon and nitrogen. Glucose is a major metabolic substrate that can be utilized by all living cells as a source of (1) bioenergetic equivalents in a form of high-energy phosphate bonds of adenosine triphosphate (ATP) and (2) carbon building blocks, which can be used to produce a variety of biomolecules. One key chemical constituent that a glucose molecule does not provide is nitrogen. Nitrogen is required for the biosynthesis of amino acids and nucleotide bases, thus, an access to a source of reduced nitrogen is critical for both unicellular eukaryotes and metazoans. Although some bacteria can reduce atmospheric nitrogen to ammonia, most species must acquire a reduced form of nitrogen from the environment. Unicellular organisms can use free ammonia (NH4+) as a source of reduced nitrogen. For example, a wild-type, or prototrophic, yeast culture can proliferate in a so-called synthetic complete medium, which consists purely of glucose as a sole carbon source and ammonia as a sole nitrogen source. A yeast cell has all metabolic enzymes to synthesize a complete spectrum of biomolecules from the materials provided.
Metazoan organisms rely on glucose as a preferred carbon source and amino acids as the primary source of reduced nitrogen. In contrast to unicellular organisms, metazoans have lost the ability to synthesize 9 out of 20 amino acids required for protein synthesis (these nine amino acids are termed “essential,” while the rest are termed “nonessential”). The amino acid glutamine, a carrier of two nitrogen atoms, is the predominant “nitrogen currency” for metazoan organisms. Because of its special importance as a reduced nitrogen source, glutamine is a “conditionally essential” amino acid for most proliferating metazoan cells.8
The significance of glucose and glutamine as key metabolic substrates for metazoan cells can be illustrated by the fact that in a mammalian organism, nearly constant levels of these two nutrients are maintained in plasma at all times: glucose levels are within the range of 4–6 mM and glutamine, the most abundant amino acid in plasma, ranges within 0.6–0.9 mM.9 These steady levels of glucose and glutamine are maintained by a combination of hormone-mediated utilization of glycogen stores in muscle and liver, de novo synthesis of glutamine and glucose, as well as the stimulation of feeding behavior.
Unicellular organisms do not have steady levels of carbon and nitrogen sources around them at all times. Instead, they rely on nutrient sensing mechanisms to assess the availability of nutrients in their environment. In yeast, for instance, glucose limitation leads to Snf1 kinase [a homolog of adenosine monophosphate (AMP)-activated protein kinase (AMPK) in mammalian cells] activation, which promotes energy production and suppresses biosynthetic processes.10 Addition of glucose to a yeast culture triggers a shift toward biosynthetic metabolism and proliferation.11, 12 When glucose is abundant, Ras1 and Ras2 GTPases, homologs of the mammalian RAS protein family, become active and promote cell cycle progression. Another signaling mediator downstream of glucose is Sch9 kinase, which is related to Akt and S6 kinases in mammals and plays a role in promoting the use of glucose for biosynthetic processes.13 Concurrently, the amino acid status of a yeast cell is measured through Gcn2 serine/threonine kinase, which is homologous to mammalian GCN2. Gcn2 is activated by uncharged tRNA, which accumulates in cells when amino acid levels are insufficient. Activated Gcn2, in turn, inhibits protein translation rates and induces Gcn4 (ATF4 in mammals), a transcription factor that controls expression of a number of enzymes involved in de novo synthesis of amino acids.14 In the absence of glucose, however, active Snf1 represses Gcn4 expression, thus coordinating the metabolism of nitrogen and carbon.15 As intracellular amino acid levels rise, they, in turn, activate TOR1/2 (mTOR in mammalian cells), which acts as a coordinator of a number of anabolic processes, including protein translation and biosynthesis of fatty acids. If nutrient sources remain plentiful, cell growth is converted into cell division and the process is repeated until the extracellular nutrient supply falls below the necessary threshold.
Cells within a metazoan organism are surrounded by ample glucose and glutamine in plasma and interstitial fluid, yet in contrast to yeast, they do not proliferate unless a proper extrinsic growth signal is present. First of all, the very access of cells to the extracellular pools of glucose and glutamine is controlled by receptor tyrosine kinases, proteins that are absent in unicellular eukaryotes. We now appreciate that stimulation by a cytokine or a growth factor, for example, directs a cell to take up nutrients, providing it with a supply chain of metabolic substrates. To this end, signals from receptor tyrosine kinases converge on PI-3 kinase/Akt, activation of which triggers rapid translocation of glucose transporters (GLUTs) from the cytosol-localized endosomal vesicles to the plasma membrane,16, 17 as well as stimulates GLUT gene expression,18 driving the entry of glucose into cells. In illustration of this paradigm, PI-3 kinase/Akt signaling facilitates glucose uptake by populations of cells that are induced to undergo proliferative expansion, for example, activated T cells mounting an immune response,19 or those induced to synthesize massive amounts of macromolecules, such as milk-producing cells of the mammary gland during lactation.20
In addition to effects on glucose transport, growth-factor-stimulated mammalian cells rapidly induce transcription of a set of immediate-early genes, one of which is the transcription factor c-Myc. C-Myc drives expression of a large set of metabolic genes and plays a role as a gatekeeper of cellular access to glutamine—the major metazoan source of nitrogen.21 To this end, c-Myc facilitates the expression of glutamine transporters, ASCT2 and SN2,22 as well as a glutaminase enzyme, GLS1, which catalyzes the breakdown of glutamine to glutamate and free ammonia.23 Through facilitation of glutamine acquisition, c-Myc also indirectly influences the uptake of essential amino acids by the cell. Essential amino acid transport into cells is coupled to the efflux of glutamine into the extracellular space.24 This mechanism allows cells to coordinate the import of essential amino acids to intracellular glutamine status.
Besides signals from receptor tyrosine kinases, cellular anchorage to the extracellular matrix is also needed to facilitate glucose uptake. Glucose consumption is suppressed in cells that are deprived of the matrix attachment even in the presence of soluble growth factors.25 This ensures that not only proliferation, but also positions of cells within the tissue are controlled on a metabolic level.
Taken together, pro-growth signaling through receptor tyrosine kinases has emerged as a control point for nutrient acquisition. Although conserved from yeast to mammals, mammalian RAS is no longer governed by glucose abundance, but by tissue-specific receptors. Similarly, mammalian TOR, while retaining its sensitivity to amino acid levels, have been harnessed by growth signaling control via Akt-dependent phosphorylation and inactivation of TSC2, a negative regulator of mTOR. Next, we will next examine how genetic alterations in growth signaling pathways influence the nutrient uptake in cancer cells.
Key Points
Nutrient abundance is sufficient to stimulate proliferation of unicellular organisms, but not of metazoan cells, which has relegated the control over the ability to access nutrients around them to extrinsic growth stimuli.
Bad table manners of cancer cells
Discovery of the fundamental differences in rates of nutrient uptake between tumors and nontransformed tissues was among the earliest in modern cancer research, predating the discovery of the oncogenes and the role of carcinogens in tumorigenesis. In the 1920s, biochemical characterization of cancer cells from peritoneal ascites and slices of normal, nonproliferating tissues by German physiologist Otto Warburg revealed that the rate of consumption of glucose by tumors far exceeds that of normal tissues.26 Some 30 years later, American pathologist Harry Eagle showed that glutamine requirements of HeLa cells in culture exceed the needs for other amino acids by 10- to a 100-fold, far surpassing the quantity needed for protein synthesis.27
On a signaling level, cellular glucose uptake is controlled through PI-3 kinase/Akt, which targets GLUTs to plasma membrane, as well as promotes their expression on a transcriptional level.17 Thus, cancer-specific alterations in PI-3 kinase/Akt signaling allow cancer cells to consume glucose constitutively. Activated RAS and c-Src can promote GLUT1 expression as well.28 To match the supply of carbon with a supply of nitrogen, tumor cells concurrently increase their uptake of glutamine. As described previously, the key driver of glutamine uptake and utilization in mammalian cells is c-Myc.22, 29 With c-Myc levels aberrantly elevated, cancer cells continuously import and utilize available glutamine for bioenergetic support, nucleotide biosynthesis, and to coordinate essential amino acid uptake and nonessential amino acid biosynthesis in support of protein production.
Tumors originate within the confines of a normal tissue, which is not suited to sustain uncontrolled cellular proliferation. Thus, tumors often lack adequate vasculature, as de novo angiogenesis within tissues is negatively regulated by tissue-specific factors.30 Capillaries that do form in response to tumor-secreted pro-angiogenic molecules tend to be underdeveloped and inefficient in delivering oxygen and nutrients to a growing tumor. As proliferation of cancer cells depletes the microenvironment around them of amino acids and glucose, growing tumors face increasingly unfavorable conditions for proliferation. One adaptation that cancer cells use to withstand nutrient limitations is autophagy—a regulated process of self-catabolism of proteins, lipids, and even whole organelles as a temporary means of sustaining viability during starvation.31 A major cellular bioenergetic sensor, AMPK, initiates the process of autophagy in response to falling ATP/AMP levels, whereas mTOR, on the other hand, acts as an autophagy inhibitor.32 The significance of autophagy for tumor growth in vivo has been demonstrated in mutant KrasG12D and Braf V600E-driven lung tumors, in which the genetic knockout of the autophagy machinery component Atg7 dramatically limits tumor cell accumulation and aggressiveness.33, 34
Although autophagy can serve as a temporary means to survive nutrient shortages and maintain cellular bioenergetics, it cannot be used as a source of new biomass. In addition to intracellular proteins, KRAS- and c-Src-transformed cells have been recently shown to recover amino acids from proteins dissolved in extracellular fluid, which are consumed via large (up to 1–2 µm in diameter) vesicles at sites of plasma membrane ruffling.35 These vesicles are called macropinosomes (from latin pino—“to drink”). Under conditions of nutrient depletion, macropinosomes are targeted to lysosomes, where engulfed cargo is degraded. This alternative, “scavenging” form of nutrient uptake allows cells to survive amino acid limitations by unlocking otherwise inaccessible amino acid stores. RAS-driven macropinocytosis is an evolutionarily ancient mechanism of acquiring nutrients. In fact, it can be a primary pathway of nutrient acquisition in slime molds, such as Dictyostelium.36 Thus, reactivation of macropinocytosis in oncogene-transformed cells can be viewed as yet another example of how cancer cells utilize evolutionarily ancient mechanisms of nutrient uptake.
Another problem that stems from the inadequacies of tumor vasculature is reduced supply of oxygen, or hypoxia. Lack of sufficient oxygen has a profound effect on cellular metabolism as it inhibits those biochemical reactions in which oxygen is consumed. One reaction that becomes inhibited in hypoxic conditions is stearoyl-CoA desaturase 1 (SCD1)-catalyzed desaturation of fatty acid carbon bonds to create unsaturated fatty acids, which are essential for proper fluidity of plasma and organelle membranes. To circumvent this problem, Ras-transformed cells were shown to be able to bypass the need for SCD1-catalyzed desaturation reaction by scavenging unsaturated lipids from the extracellular environment.37
Key Points
Oncogene signaling allows cancer cells to use conventional metabolic sources, glucose and glutamine, in an unrestricted manner, as well as enables cells to access unconventional sources of biomolecules.
The traveling electrons
Most biochemical reactions within living cells are energetically unfavorable and require catalysis by specific metabolic enzymes. Metabolic reactions can be divided into two types: (1) catabolic—in which complex biochemical substrates are degraded to simpler constituents and (2) anabolic—in which complex biomolecules, such as amino acids, fatty acids, and nucleotides, are produced from simpler constituents. Catabolic processes involve reactions in which substrates undergo oxidation. Electrons derived from these oxidative steps are captured in a form of hydride anions (:H−), which are carried by specialized enzyme cofactors NAD+ and FAD (NADH and FADH2 in their reduced forms), or NADP+ (NADPH in its reduced form). The chemical energy of electrons from NADH and FADH2 is then utilized by a series of enzyme complexes of the electron transport chain, which is located at the inner membrane of mitochondria. NADH/FADH2-derived electrons travel down the electron transport chain until they react with oxygen to produce water. The chemical energy of electrons that are being passed along the electron transport chain is used to create a proton gradient between the mitochondrial matrix and the mitochondrial intermembrane space. The resulting build-up of electrochemical potential across the inner mitochondrial membrane, in turn, fuels the activity of ATP synthase, a supramolecular complex that regenerates high-energy phosphate bonds of ATP from ADP. The coupling of electron transport to ADP conversion to ATP is termed oxidative phosphorylation.
In living cells, the ratio of ATP to ADP provides the thermodynamic driving force for a myriad of energetically unfavorable cellular processes, as well as for protein and lipid phosphorylation by kinases—a versatile mode of regulating functions of a variety of proteins and transmitting signals within a cell.
The maintenance of an electrochemical proton gradient through the electron transport chain is dependent on the availability of oxygen—the terminal acceptor of electrons, and ADP—a substrate for the ATP synthase. An excessive influx of electrons from NADH/FADH2 can deplete the supply of either of these, stopping the oxidative phosphorylation. Consequently, electrons that become stalled in the electron transport chain can be lost over time from their intramembrane carriers, reacting with the aqueous environment of the cell to produce reactive oxygen species (ROS). ROS are avid oxidants, which readily react with a wide spectrum of biomolecules—including lipids, proteins, and nucleotides. To control their redox state, cells produce a variety of ROS-neutralizing molecules, or antioxidants. However, when ROS become elevated beyond the cellular antioxidant capacity, they can cause widespread damage to cellular organelles and DNA.
As noted previously, most catabolic reactions couple the oxidation of their substrates to the reduction of NAD+ and FAD to NADH and FADH2. In contrast, a few metabolic reactions result in a transfer of electrons to a structurally related cofactor, NADP+, reducing it to NADPH. Whereas NADH and FADH2 are mainly used for energy production, NADPH replenishes the cellular antioxidant pool by sustaining the reduced state of glutathione and related sulfhydryl-containing molecules. In addition, the reducing power of NADPH is used in biosynthesis of complex biomolecules, such as fatty acids and nucleotides, which require sequences of reductive reactions. How does a cell choose to prioritize one class of such reactions over others? As it turns out, the allocation of substrates into processes that support bioenergetics versus those with biosynthetic outcomes is coordinated by many of the same pro-growth signals that promote the uptake of biochemical substrates by cells in the first place.
Key Points
A living cell maintains its viability by oxidizing biochemical substrates acquired from the environment. Catabolic reactions produce simple building blocks, which can be consecutively utilized in biosynthetic reactions. In addition, catabolic reactions serve as sources of reducing power to fuel cellular energy production, enable biosynthetic reactions, and bolster the antioxidant defense.
Warburg effect—how to play safe while looking sloppy
Glucose catabolism takes place in two cellular compartments: its first stage, glycolysis, occurs in the cytosol, while the second stage is localized to mitochondria and involves a series of reactions of the tricarboxylic acid cycle (TCA cycle, which is also called citric acid or Krebs cycle). Phosphorylation of glucose by a hexokinase (HK) to produce glucose 6-phosphate is the first committed step of glycolysis (Figure 1a). The HK reaction prevents the exit of glucose back into the extracellular space. A second committed step of glycolysis is an irreversible phosphorylation catalyzed by phosphofructokinase enzyme (PFK1). Along with targeting the GLUT1 transporter to the plasma membrane, Akt potentiates the action of both HK38 and PFK1,39 thus acting as a coordinator of glucose progression through glycolysis. In fact, the ability of Akt to suppress apoptosis in response to growth factor withdrawal is dependent on glucose;40 moreover, overexpression of GLUT1 and HK alone is sufficient to suppress growth factor withdrawal-induced cell death.39 PFK1 activity in cancer cells is further enhanced by frequent overexpression of PFKFB3,41 an enzyme that produces fructose 2,6-bisphospate, an allosteric activator of PFK1.
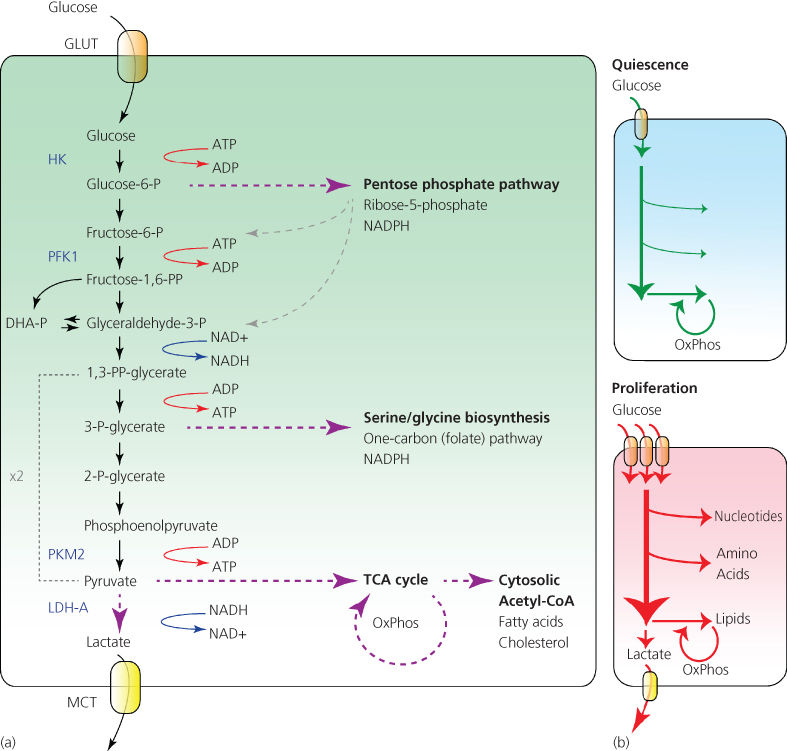
Figure 1 Glycolysis. (a) Catabolism of one glucose molecule via glycolysis yields two molecules of pyruvate, which can then be transported to the mitochondria and used as a source of acetyl-CoA for the TCA cycle. Alternatively, pyruvate can be reduced to lactate and excreted out of the cell. Besides its bioenergetic uses, glycolysis also provides biosynthetic power to cells. Glucose-6-phosphate can be utilized in the PPP to generate NADPH and ribose 5-phosphate for nucleotide biosynthesis; or, its intermediates can be shunted back into glycolysis (shown by gray dashed arrows). In addition, 3-phosphoglycerate can be converted to serine, a carbon donor for the folate (one-carbon) cycle. Folate cycle generates NADPH, as well as provides one-carbon units for the biosynthesis of nucleotide bases and for methylation reactions. GLUT, glucose transporter, HK, hexokinase, PFK1, phosphofructokinase 1, PKM2, pyruvate kinase M2, LDH-A, lactate dehydrogenase A, MCT, monocarboxylate transporter, OxPhos, oxidative phosphorylation. (b) Quiescent cells consume moderate amount of glucose and preferentially catabolize it to pyruvate to fuel oxidative phosphorylation in mitochondria. In proliferating cells, growth factor signaling or constitutively active oncogenes facilitate the increase in glucose uptake. In contrast to a quiescent cell, the majority of glucose consumed by a growth-stimulated cell goes into biosynthetic pathways to produce nucleotides, amino acids, and lipids—or is expelled as lactate.
Downstream of glucose capture and degradation, the various intermediates of glycolysis and the TCA cycle provide the backbones of essentially all the amino acids, acyl groups, and nucleotides a cell can synthesize de novo to support macromolecular synthesis. When it was discovered that oncogenic mutations resulted in flooding the cell with glucose that can maintain a supply of these precursors, it became clear why the Warburg effect was selected for during tumorigenesis. A cell engaged in excessive glucose uptake becomes resistant to apoptosis, has virtually unlimited supply of macromolecular precursors, and simply secretes any carbon in excess of its needs by converting the end product of glycolysis, pyruvate, into lactate, which is expelled from the cell. This form of gluttony exhibited by tumors harboring mutations that increase glucose uptake became known as aerobic glycolysis—to distinguish it from the well-established increase in glucose uptake exhibited by normal cells when depleted of oxygen, or anaerobic glycolysis.
Recently, it has been shown that aerobic glycolysis is not restricted to tumor cells. When stimulated with growth factors, normal cells too, begin to display Warburg-like metabolism.42 The discovery that the Warburg effect is a growth-factor-driven phenomenon and not a peculiar “abnormality” of cancer cells warranted exploring whether it may be in fact, a part of a metabolic reprogramming aimed at increasing the cellular capacity for biosynthesis. Thus, it is now becoming appreciated that maximization of the bioenergetic yield from glucose catabolism would run contrary to the biosynthetic priorities of a growth-factor-stimulated cell.43
How can glycolysis help biosynthesis? There are several ways through which glycolytic intermediates can act as substrates for producing both biosynthetic precursors and NADPH (Figure 1a). One such pathway is the pentose phosphate pathway (PPP), which begins with glucose 6-phosphate. Glucose 6-phosphate is oxidized to produce two NADPH molecules, and after a series of interconversions, it produces ribose 5-phosphate—a structural component of nucleotides. Significantly, the PPP enzyme transketolase-like 1 (TKTL1) is required for growth of a number of cancer cell lines and is predictive of poor prognosis in patients.44
Another key branch point of glycolysis is at the level of 3-phosphoglycerate. In a series of three consecutive reactions, 3-phosphoglycerate is converted to the amino acid serine. As metabolic studies demonstrate, serine biosynthesis is massively upregulated in tumors beyond the amount necessary for protein biosynthesis.45 Moreover, PHGDH gene, encoding the enzyme that catalyzes the limiting step in serine biosynthesis, is frequently amplified in breast cancer and in melanoma, predicts tumor aggressiveness and is required for tumor growth in vivo.46, 47 Why do tumors produce so much serine? Serine is not only used as a building block in protein translation, but is also a key substrate in a so-called one-carbon, or folate cycle, where a serine-donated carbon is passed on to a tetrahydrofolate (THF) molecule. The folate cycle involves a complex series of oxidative–reductive transformations, which involve serine-donated methyl group, and is indispensable for a number of processes that affect cellular growth and proliferation.48 In particular, serine-derived carbon is used as (1) a carbon donor for purine synthesis; (2) a donor of methyl groups onto S-adenosylmethionine (SAM), a substrate for histone and DNA methyltransferase enzymes, and (3) a donor of reducing power for the production of NADPH. As a recent study has demonstrated, up to 50% of all cellular NADPH may be generated via one-carbon pathway.49 Taken together, diversion of glycolytic intermediates into the pentose phosphate shunt and into the serine biosynthesis pathway both provide building blocks for nucleotide generation as well as produce NADPH.
How does a cell regulate the rate at which glycolytic intermediates are diverted into biosynthetic pathways? Exploration of the interplay between growth factor signaling pathways and metabolism have led to a seminal discovery that has shed light on this question. Specifically, tumor cells express an M2 splice variant of pyruvate kinase M (called PKM2), while their normal counterparts preferentially express the M1 variant.50 Pyruvate kinase is a rate-limiting enzyme that catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate (Figure 1a). Intriguingly, the PKM2 isoform is a less active form of pyruvate kinase than PKM1. Moreover, PKM2, but not PKM1, is susceptible to further inhibition via binding to phosphorylated tyrosine residues through its unique C-terminal sequence.51 Thus, the activity of PKM2 becomes dampened even further in response to increased tyrosine kinase signaling. Replacement of PKM2 with a PKM1 isoform reduces the tumorigenic potential of cancer cells, promotes oxygen consumption, and inhibits production of lactate.50 The lower rate of catalysis by PKM2 creates a “traffic jam” at the level of PEP, facilitating the entry of accumulated glycolytic intermediates into the two pathways described previously.52 Reciprocally, products downstream of both PPP (succinylaminoimidazolecarboxamide ribose 5′-phosphate or SAICAR)53 and serine biosynthetic pathway (serine itself)54 act as allosteric activators of PKM2, which allows a cell to balance the biosynthetic and bioenergetic uses of glycolysis. Besides its function as a gatekeeper of glycolysis, PKM2 also promotes glucose utilization at the level of gene expression. Thus, in EGF-treated cells, PKM2 becomes phosphorylated by Erk1/2 and is translocated to the nucleus, where it directly controls expression of GLUT1 and lactate dehydrogenase A (LDH-A) genes, further contributing to the Warburg effect.55
Increased glycolytic flux allows the growing cell to dynamically regulate the production of diverse biosynthetic intermediates, at the same time secreting any surplus of carbon out of the cell in the form of lactate (Figure 1b). The previous findings help explain why growth signaling enhances glucose consumption and its entry into glycolysis, but why is glycolysis-derived pyruvate preferentially converted to lactate as opposed to entering the TCA cycle? One explanation of this phenomenon is that a cell with a high rate of glucose consumption must safeguard itself against the overproduction of NADH/FADH2 in the TCA cycle.56 Indeed, a surplus of electron donors would quickly deplete available ADP, overload the electron transport chain, and overproduce ROS. To prevent this from happening, both NADH and ATP act as allosteric inhibitors of the mitochondrial gatekeeper enzyme PDH, attenuating the production of acetyl-coenzyme A (CoA) from pyruvate and ensuring that the electron transport chain does not produce more ATP than a cell can use at the moment.
Besides being allosterically controlled by ATP and NADH, PDH is also negatively regulated via phosphorylation by pyruvate dehydrogenase kinase 1 (PDK1). Expression of PDK1 is induced when cellular production of electron donors for the electron transport chain exceeds the amount that can be assimilated by the available oxygen. Specifically, PDK1 transcription is upregulated by HIF1α, a transcription factor induced under these conditions.57, 58 Concurrently, HIF1α activates transcription of LDH-A, re-routing the flux of carbon away from the TCA cycle and toward lactate production.59 Even though HIF1α is classically activated by oxygen deficit, transformed cells exhibiting excess glycolysis induce HIF1α even in normoxic conditions.60 Nor is HIF1α the only transcriptional activator of PDK1. Indeed, another commonly activated pathway in cancer, the Wnt/β-catenin pathway, has been shown to promote the Warburg effect by upregulating the expression of PDK1, as well as the lactate transporter MCT1.61
A damaging effect of an unrestricted pyruvate catabolism has been recently proposed as a metabolic mechanism of oncogene-induced senescence (OIS). OIS is a phenomenon in which overexpression of mutant oncogenes, such as RAS or BRAF, triggers growth arrest, overproduction of ROS, and DNA damage response.62 Interestingly, the onset of OIS is dependent on PDH activity. To this end, the modulation of the PDH activity by expressing PDK1 or inhibiting PDH phosphatase PDP2 was found to facilitate cellular escape from OIS.63
A decade ago, it was discovered that the role of mitochondria in the successful evolution of the eukaryotic cells depended more on their contributions to macromolecular biosynthesis than to ATP production. In an evolutionary sense, mitochondria became essential organelles because they supplied a variety of precursors for biosynthetic reactions. In what is referred to as cataplerosis, TCA cycle intermediates exit the mitochondria to be used as precursors for a variety of anabolic reactions—including biosynthesis of sugars, nonessential amino acids, and fatty acids. In addition, mitochondria support the synthesis of iron–sulfur (Fe–S) clusters, which are central for the function of a variety of mitochondrial as well as cytosolic enzymes.
Cytosol-localized biosynthesis of fatty acids and cholesterol is particularly essential for proliferating cells. A universal precursor for fatty acid and cholesterol biosynthesis is acetyl-CoA, but because the bulky CoA cannot cross the mitochondrial membrane, a cell cannot directly access the mitochondrial pool of acetyl-CoA for use in biosynthetic reactions. Instead, the TCA intermediate citrate is shuttled out of the mitochondria and into the cytosol, where it is cleaved by ATP-citrate lyase (ACL), regenerating acetyl-CoA (Figure 2). ACL is essential for tumorigenesis, and its inhibition suppresses tumor growth in mice.64, 65 Notably, ACL enzyme becomes activated following Akt induction,66 so that Akt not only enables glucose uptake and catabolism, but also diverts carbon flux away from mitochondria and toward fatty acid biosynthesis.
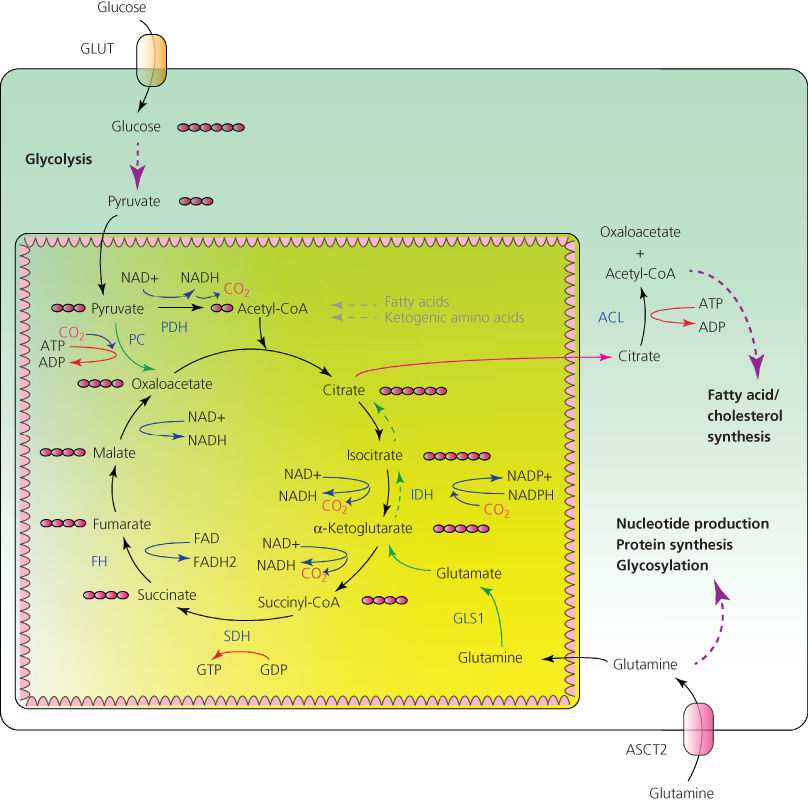
Figure 2 Carbon metabolism in mitochondria. Glycolysis-derived pyruvate serves as a donor of acetyl-CoA for the tricarboxylic acid (TCA) cycle. Cataplerotic efflux of citrate into the cytosol (shown by a maroon arrow) allows maintaining the extramitochondrial pool of acetyl-CoA for fatty acid and cholesterol biosynthesis. Glutamine and pyruvate function as anaplerotic substrates to restock the TCA cycle intermediates (shown by solid green arrows). In conditions when acetyl-CoA is limiting, citrate can be produced from the reductive carboxylation of glutamine-derived α-ketoglutarate (shown by dashed green arrows). Pink circles represent a number of carbon atoms in a molecule. GLUT, glucose transporter, PDH, pyruvate dehydrogenase, PC, pyruvate carboxylase, IDH, isocitrate dehydrogenase, SDH, succinate dehydrogenase, FH, fumarate hydratase, ACL, ATP-citrate lyase, GLS1, glutaminase, ASCT2, glutamine transporter.
Key Points
Pro-growth signals facilitate the uptake of glucose and remodel the glycolytic flux to satisfy the increased biosynthetic demands of a growing cell.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


