FIGURE 8.1 A graphic representation of Warburg’s experiment is shown. Most normal tissues produce lactate in large quantities only when oxygen (O2) is absent. In contrast, cancer cells tend to make lactate regardless of O2 availability. This tendency of cancer cells to metabolize glucose to lactate (aerobic glycolysis) is often referred to as the Warburg effect.
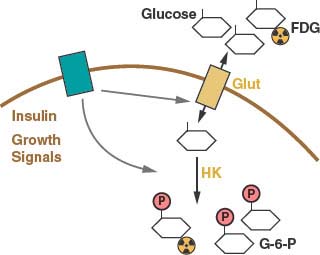
FIGURE 8.2 Glucose uptake is controlled in mammalian cells by the presence of glucose transporters on the cell surface (Glut). These transport proteins allow the diffusion of glucose across the plasma membrane where it is phosphorylated by the enzyme hexokinase (HK) and trapped in the cell. Glucose transporter expression is controlled by insulin signaling in insulin responsive tissues. Glucose uptake is also regulated by cell growth signals. A positron emission tomography (PET) scan can be used to monitor glucose uptake in the clinic. This assay uses the positron-emitting fluorine-18 (18 F)-conjugated glucose analogue fluoro-2-deoxyglucose (FDG), which can be phosphorylated by hexokinase and trapped in the cell but cannot be metabolized further.
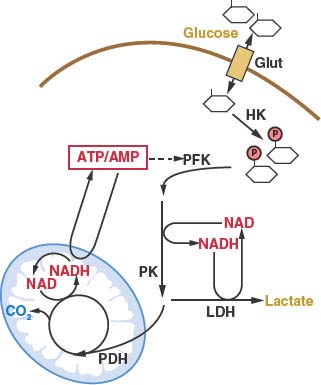
FIGURE 8.3 Cellular glucose metabolism is regulated at several steps. Uptake of glucose is regulated by glucose transporter expression (Glut). Glucose metabolism by glycolysis classically is considered to be regulated at three enzymatic steps: these include the reactions catalyzed by hexokinase (HK), phosphofructokinase (PFK) and pyruvate kinase (PK). The PFK step in glycolysis is a major point of regulation. One major allosteric input to PFK activity is the adenosine triphosphate: adenosine monophosphate (ATP:AMP) ratio. PFK activity and glycolysis are activated when the ATP:AMP ratio is low and inhibited when the ATP:AMP ratio is high. Glycolysis is also controlled by the availability of the cofactor nicotinamide adenine dinucleotide (NAD). NAD is reduced to NADH at the glyceraldehyde-3-phosphate dehydrogenase step of the pathway, and NADH must be recycled back to NAD to allow continued glycolysis. NADH can be converted to NAD by lactate dehydrogenase (LDH)-mediated conversion of pyruvate to lactate. NAD regeneration can also be coupled to the tricarboxylic acid (TCA) cycle and mitochondrial oxidative phosphorylation. Entry of pyruvate into the TCA cycle is controlled by pyruvate dehydrogenase (PDH) and allows the complete catabolism of glucose to carbon dioxide (CO2) and maximum ATP production.
Once trapped in cells, glucose can be metabolized via glycolysis to generate pyruvate in the cytosol.11 The rate of glycolysis is controlled by glucose flux into cells, cofactor availability, and the activity of glycolytic enzymes (Fig. 8.3). Both the phosphofructokinase and pyruvate kinase steps of glycolysis are highly regulated and have been implicated in the control of tumor cell metabolism (Figs. 8.3 and 8.4).14–16 Another major determinant of glucose metabolism by glycolysis is the availability of oxidized nicotinamide adenine dinucleotide (NAD) to serve as the electron acceptor for the conversion of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate. This reaction, catalyzed by glyceraldehyde-3-phosphate dehydrogenase reduces NAD to NADH (reduced nicotinamide adenine dinucleotide). Because the size of the NAD/NADH cofactor pool in cells is small relative to the flux of glucose through glycolysis, continued cycling of NADH back to NAD is critical to permit continued glycolysis (Fig. 8.3). NADH can be reoxidized to NAD through a series of reactions that shuttle reducing equivalents into mitochondria for use in oxidative phosphorylation. This process is coupled to the further metabolism of pyruvate in the mitochondrial tricarboxylic acid (TCA) cycle and can result in the generation of large amounts of ATP. ATP is used as a source of free energy for cells to enable otherwise unfavorable biochemical processes. Mitochondrial oxidative phosphorylation requires the presence of oxygen (O2) as the final acceptor of electrons from NADH and therefore is also referred to as aerobic respiration. Aerobic glycolysis also generates ATP; however, the ATP yield per molecule of glucose is much less than for aerobic respiration. The metabolism of glucose to pyruvate without mitochondrial respiration requires the enzyme lactate dehydrogenase (LDH) to produce lactate and regenerate NAD from NADH.
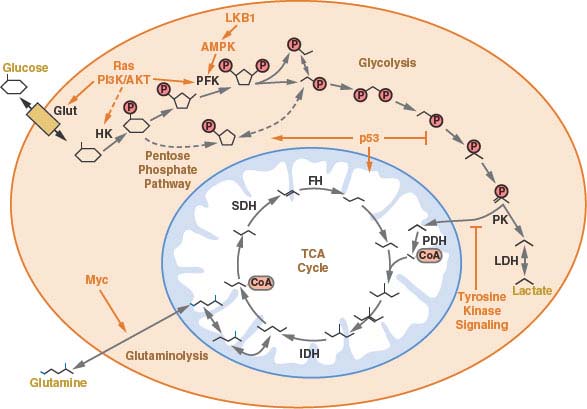
FIGURE 8.4 A schematic of central carbon metabolism is presented to show how glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway, and glutamine metabolism are interconnected in cells. The major points of enzymatic regulation, along with the enzymes discussed in the text that have been demonstrated to be important in cancer, are shown for orientation within the pathways, Glut, glucose transporter; HK, hexokinase; PFK, phosphofructokinase; PK, pyruvate kinase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; IDH, isocitrate dehydrogenase; SDH, succinate dehydrogenase; FH, fumarate hydratase. The site of regulation within these pathways by some of the major oncogenes and tumor suppressor genes is also shown.
Several hypotheses for why aerobic glycolysis appears to be selected for in cancer cells have been proposed. Warburg hypothesized that cancer cells develop a defect in mitochondria that leads to impaired aerobic respiration and a subsequent reliance on glycolytic metabolism.2 Although mutations in mitochondrial enzymes have been implicated in a subset of cancers (discussed in detail later in the chapter), subsequent studies demonstrated that mitochondrial function is not impaired in the majority of cancer cells.17 Nevertheless, despite his hypothesis being incorrect, Warburg’s original observation has held true with numerous reports describing that despite normally functioning mitochondria many cancer cells preferentially metabolize glucose via aerobic glycolysis.4,18
The growth of solid tumors is limited by the presence of an adequate blood supply to deliver oxygen and nutrients to support cell metabolism. Therefore, angiogenesis is an important process for tumor growth, and targeting this process has been successful for cancer therapy.19 Because many tumors are characterized by inefficient angiogenesis, cells must survive periods of relative hypoxia and nutrient deprivation during tumorigenesis.5 Therefore, it has been proposed that the relative hypoxia of tumors selects for glycolytic metabolism.3 However, aerobic glycolysis is observed at the earliest stages of tumorigenesis. It is a characteristic feature of leukemia and lung cancers that arises under conditions of normal to high oxygen tensions and is found in normal rapidly proliferating tissues during embryogenesis and immune responses.1 Therefore, it is likely that aerobic glycolysis provides another benefit to cancer cells that is selected for during tumorigenesis.1 Aerobic glycolysis may still facilitate tumor survival during periods of hypoxia. The same pathways that regulate angiogenesis also promote aerobic glycolysis, suggesting that important connections between these two processes exist.20,21
The selective pressure for aerobic glycolysis in cancer cells may be related to the reprogramming of metabolism to accommodate rapid cell division. Cells metabolize glucose for purposes other than generating ATP.1 Intermediates derived from the metabolism of glucose are used in other metabolic pathways in cells and ultimately provide much of the carbon necessary to produce biomass. The production of nucleic acids, amino acids, lipids, and carbohydrates needed to duplicate all the components of the dividing cell is the major metabolic requirement that distinguishes rapidly proliferating cancer cells from most normal cells. If glucose is completely catabolized to carbon dioxide (CO2), as occurs during oxidative metabolism, there are no metabolic intermediates available for biosynthetic reactions. Thus, aerobic glycolysis may reflect how metabolism is altered to permit anabolic metabolism.1 Indeed, many micro-organisms grow by fermentation when nutrients are abundant and display a metabolic phenotype analogous to aerobic glycolysis.22
ENERGETICS OF CELL PROLIFERATION
There is evidence that the rate of aerobic glycolysis, including the accompanying increased rate of glucose utilization, is not elevated in cancer cells solely to satisfy ATP demand. It has been suggested that glycolysis in tumors cells is limited by the rate of ATP consumption.23,24 Cancer cells must balance the catabolism of nutrients to generate ATP with other metabolic needs to allow net biosynthesis and cell proliferation.1 To produce a daughter cell, a proliferating cell must replicate the genome, duplicate the ribosomes and protein synthesis machinery, generate new organelles, and synthesize de novo enough lipids to duplicate cellular membranes. This imposes a large requirement of new nucleic acids, amino acids, and lipids for cell proliferation. While ATP hydrolysis provides free energy for many biosynthetic pathways, there are additional requirements to carry out these anabolic reactions. Synthesis of lipids and nucleic acids both require specific metabolite precursors and reducing equivalents provided by nicotinamide adenine dinucleotide phosphate (NADPH) (Fig. 8.5). In fact the need for NADPH and carbon skeletons on a molar basis exceeds the requirement for ATP in many biosynthetic reactions.1 Aerobic glycolysis may allow cancer cells to balance the various metabolic requirements of proliferation.
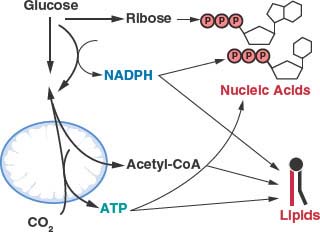
FIGURE 8.5 The generation of macromolecules requires several products of central carbon metabolism. In addition to adenosine triphosphate (ATP), the synthesis of nucleic acids and lipids both require reducing equivalents provided by nicotinamide adenine dinucleotide phosphate (NADPH) and carbon precursors that branch from different points of core metabolic pathways. Cancer cells must balance the production of specific macromolecular precursors such as ribose and acetyl-coA with the generation of enough NADPH and ATP to support proliferation.
Cancer Cells Can Metabolize Nutrients Other Than Glucose
Although tumor cell metabolism is adapted to facilitate anabolic metabolism for rapid proliferation, it is not clear why many cancer cells excrete lactate. Each lactate excreted wastes three carbons that might otherwise be recycled to fulfill some need in building a new cancer cell. It has been hypothesized that the excretion of lactate, which accompanies aerobic glycolysis, may enable faster proliferative metabolism.1 In the early stages of tumorigenesis, cancer cells are not limited for nutrients. Cells that incorporate nutrients into biomass most efficiently will proliferate faster. Lactate production may provide other advantages to a tumor as well. Acidification of the tumor microenvironment has been shown to promote invasion and metastasis.3 Lactate can also act as a nutrient for some cells in the tumor.25 As tumors grow, cells in the less-well vascularized regions of a tumor can utilize lactate from neighboring cells as a carbon source to survive periods of cell stress.26
Despite an increased reliance on aerobic glycolysis, most tumor cells continue to metabolize at least some nutrients by oxidative phosphorylation. In fact, it is likely that in some cancer cells oxidative phosphorylation continues to generate much of the ATP required for biosynthetic reactions and cellular housekeeping functions.4 Attempts to quantitate ATP production experimentally in cancer cells have estimated that 80% of the ATP generated is derived from oxidative pathways, while 20% comes from glycolysis,27 and others report that up to 50% of the ATP in cancer cells can be derived from oxidative phosphorylation.28
Glucose is not the only substrate used by cancer cells for oxidative phosphorylation.27,29 In fact, some human cancers do not demonstrate elevated glucose uptake. It has been proposed that at least some of these cancers may be dependent on the amino acid glutamine as a primary source of carbon. Glutamine can be metabolized via two transamination reactions to the TCA cycle intermediate α-ketoglutarate (Fig. 8.4), and glutamine is required for proliferation of many cancer cells.30 In addition to glucose and glutamine, other nutrients have been shown to be important in some tumors. Several clinical studies have demonstrated that some human cancers show increased uptake of acetate by positron emission tomography (PET) scan using carbon-11 (11C)-acetate as a tracer (see below).31 Acetate can be converted to acetyl-coA and serve as a precursor for lipid synthesis.32 When production of lipids from glucose is blocked by inhibiting ATP citrate lyase, an enzyme required to convert glucose to cytosolic acetyl-coA, acetate can completely rescue lipid synthesis and cell growth. Acetate is not thought to be a major nutrient available to cancer cells in patients, but may reflect an increased dependence of some cancer cells on lipid metabolism. Other enzymes involved in lipid metabolism have been linked with cancer progression,33,34 however, the exact mechanisms by which lipid metabolism promotes malignancy are not clear.
Nutrients such as amino acids and iron are also important for some tumors.35,36 Cachexia, or the loss of body mass that cannot be reversed by increased nutrition, is associated with the late stages of some cancers. Cachexia is poorly understood, but in cancer patients it is characterized by the loss of adipose tissue and muscle mass.37 This may reflect a derangement in whole body metabolism to supply specific nutrients to the cancer. Understanding how nutrients other than glucose contribute to cancer biology remains an active area of investigation.
IMAGING CANCER METABOLISM IN PATIENTS
The characteristic increased glucose uptake of cancer cells has been exploited to image cancer in the clinic. The glucose analogue 2-deoxyglucose is permeable to glucose transporters and trapped in cells when phosphorylated by hexokinase (Fig. 8.2). The 2-deoxyglucose-6-phosphate is unable to be further metabolized,38,38 and thus 2-deoxyglucose-6-phosphate accumulation can be used to assess the rate of glucose uptake into cells. By conjugating the positron emitting isotope fluorine-18 (18F) to 2-deoxyglucose, the uptake of glucose can be measured in patients using PET.40 18F-fluro-2-deoxyglucose (FDG)-PET is used widely in the clinic to visualize tumors. The current use of FDG-PET is primarily as a staging tool for cancers, however, it is also sometimes used to characterize lesions observed by other imaging modalities. FDG-PET is also increasingly being used as a marker of response to therapy (Fig. 8.6). At least for some treatments, there is mounting evidence that decreased uptake of FDG by PET scan following therapy is a predictor of clinical efficacy.41,42
In the clinic, FDG-PET scan can be used to classify tumors. Although many tumors are visible by FDG-PET scan, some tumors do not display elevated FDG uptake. This illustrates that different tumors can display distinct metabolic phenotypes related to their genetic background or site of origin. As discussed above, some tumors rely on nutrients other than glucose. For instance increased 11C-acetate uptake has been observed by PET in prostate tumors that do not demonstrate elevated signal on FDG-PET scan.43 However, increased uptake of 11C-acetate is also seen in some benign prostate conditions, so whether 11C-acetate uptake defines a characteristic of some prostate cancers or reflects the underlying biology of the prostate gland remains to be determined.44 New methods to image tumor metabolism by PET or magnetic resonance imaging (MRI) are being developed. If successful, such efforts will better define distinct metabolic phenotypes in patient tumors.
GENETIC EVENTS IMPORTANT FOR CANCER INFLUENCE METABOLISM
Human cancer occurs as a consequence of genetic events that promote the inappropriate proliferation of cells.45 These events lead to the expression of oncogenes or the loss of tumor suppressor genes that contribute to tumor formation and progression. Although alterations involving specific oncogenes or tumor suppressor genes are hallmarks of specific malignant phenotypes, many of these genetic events are found in numerous types of cancer. These genetic changes occur in diverse cellular signaling and transcriptional pathways, and it is unclear how the various mutations converge to allow inappropriate cell growth and proliferation. One common downstream consequence of these genetic changes is altered cellular metabolism.
Efforts to understand the predisposition to malignancy displayed by von Hippel-Lindau (VHL) syndrome patients identified a link between cancer genetics and the regulation of cell metabolism.46 VHL syndrome results when patients inherit one mutated copy of the VHL gene, leading to a spectrum of benign and malignant tumors including clear cell carcinoma of the kidney. In these patient’s tumors, the normal VHL allele is lost, consistent with VHL acting as a tumor suppressor gene. Subsequent work demonstrated that the VHL gene product is also commonly lost in sporadic renal clear cell carcinomas. Although the exact mechanism by which VHL loss leads to renal cell carcinoma and other tumors is not known, loss of VHL has a profound impact on metabolic gene regulation, and these effects are thought to be important in the pathogenesis of these cancers.
The VHL gene encodes the substrate recognition component of a ubiquitin E3 ligase.46 As an E3 ligase, the VHL protein-containing complex facilitates the transfer of ubiquitin to specific target proteins to effect their degradation. Among the targets of the VHL protein is the hypoxia-inducible transcription factor-1α (HIF-1α) (Fig. 8.7). HIF-1α levels are regulated by oxygen tension.21,47 In the presence of oxygen HIF-1α is hydroxylated. The hydroxylated form of HIF-1α is recognized by VHL, leading to ubiquitination and degradation of the protein. When oxygen is absent, or when VHL is lost, HIF-1α protein accumulates, dimerizes with HIF-1α, and promotes the transcription of a number of hypoxia inducible genes. These genes include factors that promote angiogenesis to improve tissue blood supply as well as glucose transporters and most of the enzymes in glycolysis. Thus, inappropriate HIF-1α activation leads to the characteristic increased glucose uptake and glycolysis that is observed in cancer. HIF-1α accumulation is a direct consequence of VHL loss, providing a link between the loss of a tumor suppressor gene and the altered metabolic phenotype of malignant cells.
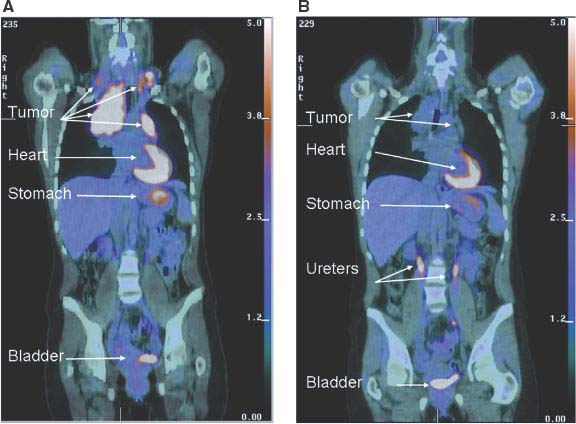
FIGURE 8.6 Early metabolic changes on 18F-fluro-2-deoxyglucose position emission tomography/computed tomography (FDG-PET/CT) are highly predictive of final treatment response. Fused coronal FDG-PET/CT images in a 44-year-old woman with stage IIA Hodgkin’s lymphoma at presentation (A) and after two cycles of ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) chemotherapy (B). The tumor seen in the mediastinum and bilateral neck shows intense avidity for the glucose analogue FDG prior to therapy consistent with increased glucose uptake in (A). Although a residual tumor mass is still seen on CT after two cycles of ABVD chemotherapy (B), it is no longer FDG-avid, and the patient has been in remission for over 2 years since completion of therapy. Normal FDG uptake and excretion is seen in the stomach, heart, and urinary tract, respectively. (Image courtesy of Tricia Locascio, Katherine Zukotynski, and Annick D. Van den Abbeele, Department of Imaging, Dana-Farber Cancer Institute.)
Increased expression of HIF-1α-regulated genes is found in many different types of cancer and correlates with poor patient prognosis.48 Expression profiling of transformed cells demonstrates that metabolic genes are among the most strongly up-regulated groups of genes.49 Loss of expression of the HIF-1α target genes correlates with response to therapy in at least some models of cancer, suggesting that these genes are important for malignant cell proliferation and survival. Importantly, increased expression of glucose transporters and glycolytic enzymes under the control of HIF-1α are seen even in non-hypoxic tumors expressing VHL.49,50 HIF-1α also drives expression of pyruvate dehydrogenase kinase, a negative regulator of the pyruvate dehydrogenase complex that catalyzes pyruvate entry into mitochondria.51,52 Thus HIF-1α expression can promote aerobic glycolysis in cancer cells.
Transcription factors other than HIF-1α can promote the expression of metabolic enzymes. Expression of ChREBP-1, mondoA, and SREBP-1 has also been shown to be important in some cancers. ChREBP-1 is a key regulator of glycolytic enzyme expression and can promote anabolic metabolism.53 MondoA is a key regulator of metabolic gene expression and coordinates glucose and glutamine metabolism.54 Enzymes involved in cholesterol and lipid metabolism are controlled by the SREBP transcription factor,55 and SREBP is induced as a result of oncogenic signaling.56 Finally, increased HIF-1α−mediated gene transcription is observed in many cancers in the absence of hypoxia or VHL loss.48 This has been attributed to increased production of HIF-1α that results from aberrant signaling downstream of phosphatidylinositol-3-kinase (PI3K), as discussed below.
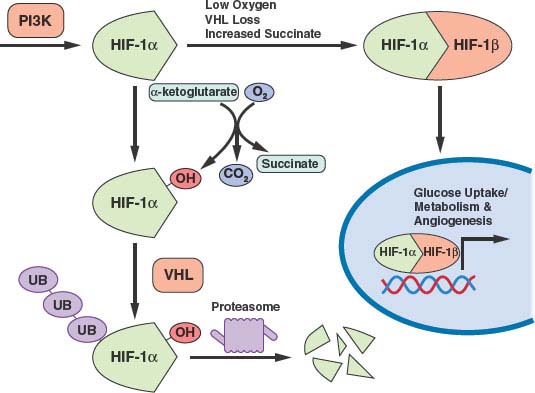
FIGURE 8.7 Transcription of many genes related to metabolism and angiogenesis is controlled by hypoxia-inducible transcription factor-1α (HIF-1α). HIF-1α levels are regulated by translational control downstream of phosphatidylinositol-3-kinase (PI3K) signaling and by the rate of proteosomal degradation. HIF-1α is marked for degradation by hydroxylation. This hydroxylation is sensitive to O2, α-ketoglutarate, and succinate levels. The von Hippel-Lindau (VHL) protein facilitates ubiquitination and degradation of hydroxylated HIF-1α. Thus, PI3K signaling, hypoxia, succinate levels, and the presence of VHL all influence transcription of HIF-1a target genes. (Figure courtesy of Brooke J. Bevis, Whitehead Institute.)
Many Genetic Drivers of Cancer Increase Nutrient Uptake
Highlighting the central role that altered cellular metabolism plays in tumor biology, one shared consequence of many genetic events that promote cancer is increased glucose uptake and metabolism. Transformation is associated with elevated glucose uptake.57,58 Activation of the PI3K signaling pathway is a common event in human cancer, and PI3K has a well-described role in glucose metabolism through its action as a proximal mediator of insulin signaling that leads to glucose uptake.59 However, even in noninsulin dependent tissues, PI3K signaling can regulate glucose uptake and utilization and appears to drive this process in many cancers.
PI3K is activated downstream of receptor tyrosine kinases and transfers a phosphate to the membrane lipid phosphatidylinositol.59,60 When phosphorylated on the 3-position (to generate phosphatidylinositol-3,4,5-triphosphate), the phosphorylated inositol species can recruit other kinases to the membrane, including the protein kinase AKT (also known as protein kinase B). Activation of AKT leads to increased expression of glucose transporters such as Glut1 and enhances the proximal steps in glycolysis by increasing hexokinase and phosphofructokinase activity (Fig. 8.4). Point mutations that result in growth-factor independent activation of PI3K are found in many human cancers, including a significant percentage of breast, ovarian, and colon cancers.60 Loss of phosphatases that degrade the phosphatidylinositol species, leading to activation of signaling downstream of PI3K, are also common events in human cancer. Loss of the phosphatidylinositol-3,4,5-triphosphate-3-phosphatase PTEN is frequently observed in human cancer, including a sizable fraction of prostate cancer, breast cancer, glioblastoma, and melanoma.60 Similarly, loss of INPP4B, a phosphatidylinsolitol-3,4-bisphosphate-4-phosphatase, is seen in breast and ovarian cancers.61
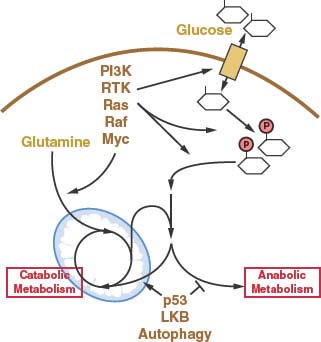
FIGURE 8.8 Many of the oncogenic events thought to drive cancer development influence metabolism by increasing glucose or glutamine uptake. Other genetic events and processes associated with cancer also influence metabolism but do not necessarily cause elevated nutrient uptake. Rather, these events appear to be involved in reprogramming of metabolism away from catabolic metabolism and promote the anabolic pathways necessary for cell growth and proliferation. Both the increase in nutrient uptake and the reprogramming of metabolism to support anabolism are required for cancer cells to proliferate. RTK, receptor tyrosine kinase.
In addition to regulating glucose uptake and capture through activation of AKT, PI3K activation also leads to increased expression of enzymes in glucose metabolism via increased production of HIF-1α.62 Activation of the mTOR kinase, an event downstream of PI3K signaling, induces expression of HIF-1α target genes at least in part through increased translation.63–65 This activation of glucose metabolism results in the propensity for tumors with PI3K activation to be FDG-avid on PET scan. Small molecules that disrupt PI3K signaling lead to decreased glucose uptake by the tumors as measured by FDG-PET, and the ability to inhibit tumor FDG uptake correlates with tumor regression in PI3K-driven animal models of cancer.61
Increased glucose uptake and metabolism are also characteristic features of tumors with activated RAS signaling. RAS activation is frequently observed across human cancers. Glucose deprivation has been shown to drive the emergence of cells harboring an activating mutation in the KRAS gene.62 This has led to the hypothesis that increased glucose uptake via Glut1 expression is a major downstream mediator of RAS signaling that contributes to tumorigenesis. In addition to increasing glucose transporter expression, RAS activation leads to increased phosphofructokinase activity and stimulates the proximal metabolism of glucose (Fig. 8.4).14 Though less well studied, activating mutations in B-Raf, a kinase downstream of RAS, also result in increased nutrient uptake.67 Mutations in receptor tyrosine kinases cause increased signaling through both the RAS and PI3K signaling pathways and also cause increased nutrient uptake.
Increased MYC expression is another frequent event in human cancer. MYC is a transcription factor, and many of the transcriptional targets of MYC include metabolic genes.50 MYC regulates the transcription of enzymes involved in glucose uptake and metabolism, including LDH, the enzyme responsible for production of lactate. MYC-dependent tumors are sensitive to LDH inhibition.68 MYC also directly and indirectly regulates the uptake of glutamine,30,69 and MYC-dependent cancer cells are particularly sensitive to glutamine withdrawal.70 Genetic alterations that lead to activation of MYC, PI3K, RAS, Raf, and receptor tyrosine kinases are among the best-understood driver events in human cancers. These seemingly disparate genetic alterations that promote cancer and occur across cancer types all lead to a converging metabolic phenotype characterized by enhanced cell autonomous nutrient uptake (Fig. 8.8).
Tumor Suppressor Gene Products Also Influence Cellular Metabolism
Many tumor suppressor gene products also regulate metabolism (Fig. 8.8). Loss of p53 function is among the most frequent genetic events in human cancer, and p53 expression influences metabolism (Fig. 8.4).71 TIGAR, a gene induced by p53 expression, redirects glucose flux from glycolysis to the pentose phosphate pathway and promotes apoptosis.72 Phosphoglycerate mutase, a glycolytic enzyme negatively regulated by p53, inhibits cell senescence.73 Excess production of reactive oxygen species also promotes cellular senescence and apoptosis, and many of the genes induced by p53 are involved in adaptation to oxidative stress and regulation of mitochondrial metabolism.74,75
The tumor suppressor LKB1 is also involved in the adaptive response to cell stress. LKB1 is a kinase that is required for activation of adenosine monophosphate (AMP)-activated protein kinase (AMPK).76–78 AMPK responds to cellular energy stress (by sensing a low ATP/AMP ratio) and initiates a signaling cascade that promotes increased ATP production and decreased ATP consumption.79 AMPK also inactivates the mTOR protein kinase that acts as an important integrator of cell growth signals with nutrient availability to regulate cell growth.80,81 In addition to AMPK, mTOR is regulated by PI3K signaling through the TSC1/TSC2 complex.82,83 TSC1 and TSC2 are tumor suppressor genes that indirectly influence metabolism via effects on mTOR signaling.84 mTOR is also regulated by amino acids such that the mTOR growth promoting activity is turned off under conditions of poor nutrient availability.85 The response to cell stress also includes the induction of autophagy to catabolize existing cellular material for energy production and removal of damaged organelles.86,87 Both mTOR signaling and nutrient stress regulate autophagy induction,88 but the role of autophagy in cancer remains unclear. The use of mTOR inhibitors is gaining increasing use in the clinic to treat various cancers, yet the relationship between mTOR, autophagy, metabolic stress, and cancer is incompletely understood.
Other genetic changes may be important to promote anabolic metabolism in cancer cells. For instance, all cancer cells appear to express the M2 isoform of the glycolytic enzyme pyruvate kinase (PK-M2).16,89 PK-M2 is normally expressed during embryonic development and is unique among the human pyruvate kinase isoforms in that it is regulated by tyrosine kinase signaling.15 This regulation of PK-M2 by growth factor signaling promotes aerobic glycolysis in cells89 and may constitute a molecular switch that allows cancer cells to metabolize glucose in a manner conducive to proliferation only when growth signals are present.
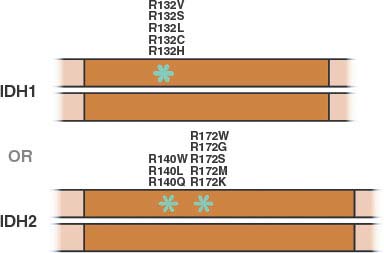
FIGURE 8.9 Mutations in isocitrate dehydrogenase-1 or -2 (IDH1 or IDH2) reported in human cancer are all monoallelic and involve a single residue in IDH1 or one of two residues in IDH2. (Figure adapted from Lenny Dang and Shengfang Jin, Agios Pharmaceuticals.)
Mutations in Metabolic Enzymes Can Lead to Cancer
Altered cell metabolism can directly contribute to transformation as mutations in metabolic enzymes can promote cancer. Inherited loss of function mutations in genes that encode proteins involved in the proper assembly or function of the mitochondrial succinate dehydrogenase complex account for the subtypes of hereditary paraganglioma.90–94 Succinate dehydrogenase (SDH) is an enzyme complex in the TCA cycle that catalyzes the oxidation of succinate to fumarate and supplies electrons to the mitochondrial electron transport chain.11 These mutations lead to decreased activity of SDH and succinate accumulation. The enzymes that carry out the oxygen-dependent hydroxylation of HIF-1α also convert α-ketoglutarate to succinate as part of their catalytic mechanism.95 Thus, accumulation of succinate leads to a decrease in HIF-1α hydroxylation, less HIF-1α degradation, and increased expression of HIF-1α target genes (Fig. 8.7). Loss-of-function SDH mutations that cause this “pseudohypoxia” account for some cases of renal cell carcinoma expressing normal amounts of VHL, supporting the notion that HIF-1α expression is an important event in some cancers.93,95 Mutations in the TCA cycle enzyme fumarate hydratase (FH), which further metabolizes the fumarate produced by the succinate dehydrogenase complex, have also been described in renal cell carcinoma, leiomyomas, and other rare cancers.95–97
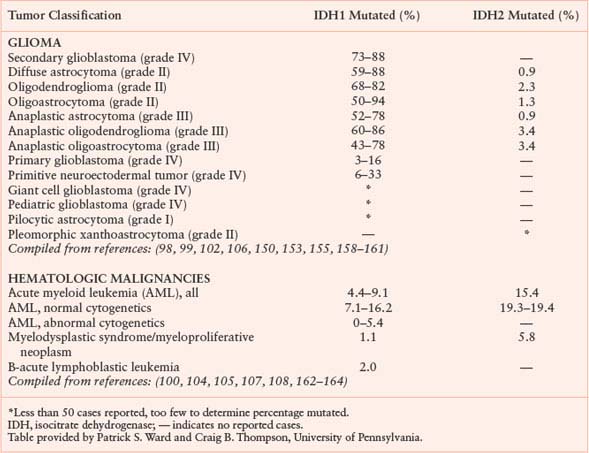
Isocitrate dehydrogenase (IDH) is another metabolic enzyme that is mutated in cancer.98–100 All reported cases of IDH mutation occur in only one allele, with preservation of wild-type enzyme expression from the other allele (Fig. 8.9).101 These mutations are frequent events in low-grade gliomas and secondary glioblastomas.98,99,102 Interestingly, IDH-mutant gliomas appear to have a better prognosis,98 suggesting that gliomas harboring these mutations represent a distinct subset of glioma with a different response to therapy or natural history. IDH mutations are also found in patients with acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) and define a distinct clinical subset of these patients without other cytogenetic abnormalities.100,103–105 IDH mutations have been proposed to be an early event in the pathogenesis of both AML and glioma.101,105,106 Sporadic mutations in other cancers have also been reported. A summary of IDH mutations in human cancer is shown in Table 8.1, and the frequency of IDH1 or IDH2 mutations in specific cancer subsets is shown in Table 8.2.
TABLE 8.1
MUTATIONS IN IDH1 AND IDH2 REPORTED IN HUMAN CANCERS
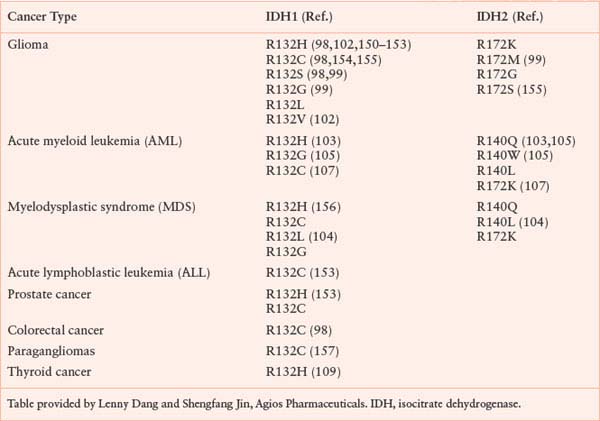
The IDH mutations associated with cancer involve point mutations in the active site of either IDH1 or IDH2.98–100,105 Other less frequent mutations involving residues in IDH1 have been reported in rare cases,105,107–109 however, whether these represent driver mutations or polymorphisms remains unclear. IDH1 mutations involve R132, and mutations in the analogous active site residue (R172) are found in IDH2. This arginine (R132 in IDH1, R172 in IDH2) coordinates the CO2 group that is lost during oxidative decarboxylation of isocitrate to α-ketoglutarate.110 The other mutated residue in IDH2 (R140) makes contacts with the same CO2 group in the IDH2 active site.105 All three mutations result in an alteration of enzyme activity that decreases the enzyme’s ability to oxidatively decarboxylate isocitrate to α-ketoglutarate, but increases enzyme activity to reduce α-ketoglutarate. In the mutant enzymes, this reduction does not lead to incorporation of CO2 to make isocitrate, but instead produces 2-hydroxyglutarate (2HG).105,107,110 Because IDH mutations are monoallelic and the neomorphic activity acquired by the mutant enzyme is slow relative to the normal wild-type enzyme in these cells, the normal interconversion of isocitrate and α-ketoglutarate is not dramatically altered, and the main consequence of these mutations is 2HG production (Fig. 8.10).105,107,110 The 2HG is produced in low levels in normal cells as an error product of various TCA cycle enzymes; however, in IDH mutant tumors 2HG accumulates to very high levels and has been linked with the pathogenesis of the disease.110 Accumulation of 2HG has been demonstrated in rare patients with hereditary loss of the enzyme that degrades 2HG,111 and gliomas have been described in a subset of these patients.112 Mutations in IDH1 have been reported to result in HIF-1α accumulation,113 however, the relationship among IDH mutation, 2HG accumulation, and HIF-1α accumulation remains unclear.
Recurrent mutations in yet another metabolic enzyme have recently been reported in colon cancer.114 These are inactivating mutations of an enzyme involved in glucosamine metabolism, and signaling involving glucosamine-based protein modifications has been suggested to be important in cancer.115 The exact mechanism by which the shunt from glucose to glucosamine impacts cellular metabolism and cancer remains to be determined.
TABLE 8.2
FREQUENCY OF IDH1 AND IDH2 REPORTED IN HUMAN CANCERS
TARGETING METABOLISM TO TREAT CANCER
The mechanisms by which cancer metabolism is regulated and how this regulation is impacted by genetic alterations known to be critical for cancer survival suggest cellular metabolic dependencies that could be exploited for cancer treatment. In fact, one of the first successful chemotherapies targeted folate metabolism. The observation that folate could enhance blood cell proliferation ultimately lead to the development of antifolates as chemotherapeutics.116 Other active chemotherapeutics target purine metabolism. Although these agents are now thought of as “cytotoxic,” they target metabolic dependencies of tumor cells and continue to play an important role in modern cancer therapy.
New therapies are being explored that directly or indirectly target cancer metabolism. A large fraction of human cancer is dependent on aberrant signaling through the PI3K/AKT pathway, and agents that target this pathway are being actively tested in the clinic.117 The growing evidence that cancer cells depend on elevated glucose metabolism suggests that targeting key metabolic control points important for aerobic glycolysis might also be effective cancer therapies. Recent evidence suggests that drugs developed to treat metabolic diseases such as diabetes may provide a benefit to patients with cancer. A number of retrospective clinical studies have found a benefit to the widely used diabetic drug metformin (Glucophage) both in the primary prevention of cancer as well as improved outcomes when used with other cancer therapies.118–120 The exact mechanism by which metformin lowers blood glucose levels is not completely understood; however, metformin decreases gluconeogenesis in the liver. Recently, metformin was discovered to be an activator of AMPK, and this effect was shown to be critical for inhibiting gluconeogenesis in the liver.121 In addition, metformin has been shown to inhibit mitochondrial complex I and impair oxidative phosphorylation in cells.122,123 Inhibition of oxidative phosphorylation and the subsequent decrease in energy charge (ATP:AMP ratio) is likely how metformin activates AMP kinase.
Recently, two separate clinical studies have shown a reduction in cancer-related mortality with metformin use.118,119 This effect appears to be independent of blood glucose level as patients with similar glucose levels treated with insulin do not derive the same benefit as patients taking metformin. Another study reported that diabetic patients on metformin had a higher pathological complete response to neoadjuvant therapy for breast cancer than diabetic patients receiving other glucose lowering therapies.120 Studies exploring metformin use for cancer therapy in numerous preclinical cancer models have suggested that these compounds may be selectively toxic to cancer cells with specific genetic backgrounds,123–125 however, these compounds may also influence cancer therapy via their systemic effects on glucose metabolism. Increased insulin-like growth factor-1 (IGF1) levels, which accompany poor glucose control, may promote cancer growth.126,127 Particularly in tumors with mutations that lead to activation of the PI3K pathway,128 at least one effect of metformin may be to decrease the effect of IGF1 on cell growth.129
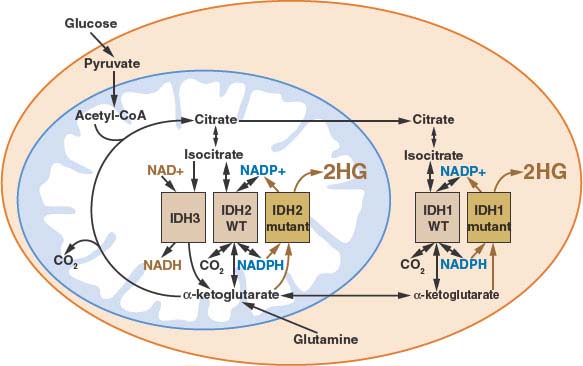
FIGURE 8.10 Human cancer associated mutations in isocitrate dehydrogenase-1 or -2 (IDH1 or IDH2) results in the neomorphic production of the metabolite 2-hydroxyglutarate (2HG). Because a wild-type version of both IDH1 and IDH2 remain even in IDH mutant cells, the normal activities of these enzymes persist despite the activity of the mutant enzyme to produce 2HG. The subcellular localization and enzymatic activities for all of the IDH isoforms is shown. (Figure courtesy of Patrick S. Ward and Craig B. Thompson, University of Pennsylvania.)
The success of FDG-PET scans as a diagnostic and predictive tool for treatment response in many cancers has lead some to suggest that targeting enzymes involved in metabolism may be an effective therapy.130 Specific therapies may be possible for patients with mutations in metabolic enzymes that contribute directly to tumorigenesis.130,131 In addition, metabolism may be sufficiently different in some cancers to target pathways such as glycolysis. 2-deoxyglucose is a competitive inhibitor of proximal glucose metabolism.38,39 Cells exposed to sufficient amounts of 2-deoxyglucose undergo growth arrest or apoptosis,132–134 and preclinical models show 2-deoxyglucose may potentiate standard cytotoxic chemotherapy.135 2-deoxyglucose can be administered to patients,136 but when given in combination with radiation therapy to glioblastoma patients at doses sufficient to limit glucose metabolism in cancer cells significant toxicity is observed.137–139 However, other agents to block glucose metabolism are being investigated. 67,130,140–146 Whether a sufficient therapeutic window exists to target glycolysis directly remains to be determined.
It is possible to alter cellular metabolism in a tumor without causing unacceptable toxicity in patients. Dichloroacetate has been used to treat lactic acidosis in non-cancer patients with rare inborn errors of mitochondrial metabolism. 147,148 Dichloroacetate reduces lactate production by inhibiting pyruvate dehydrogenase kinase (PDK),149 a negative regulator of the mitochondrial pyruvate dehydrogenase complex (PDH). PDH catalyzes the first step in mitochondrial metabolism of pyruvate (Fig. 8.4).11 Thus, PDK inhibition that leads to activation of PDH diverts pyruvate away from lactate production. Because lactate production is the hallmark of aerobic glycolysis, it has been proposed that dichloroacetate might be a useful agent to target cancer metabolism.148,149 Dichloroacetate is tolerated by cancer patients at dosages that can alter mitochondria in patient tumor samples. Studies are ongoing to understand the impact of these metabolic alterations on tumor biology and clinical outcome.
Selected References
The full list of references for this chapter appears in the online version.
1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029.
2. Warburg O. On the origin of cancer cells. Science 1956;123:309.
3. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 2004;4:891.
4. Deberardinis RJ, Lum JJ, Hatzivassiliou G, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008;7:11.
5. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell 2008;134:703.
7. Warburg O, Posener K, Negelein E. Ueber den Stoffwechsel der Tumoren. Biochemische Zeitschrift 1924;152:319.
12. Mathupala SP, Ko YH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta 2010;1797:1225.
17. Weinhouse S. The Warburg hypothesis fifty years later. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol 1976; 87:115.
19. Folkman J. Angiogenesis. Annu Rev Med 2006;57:1.
20. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer 2008;8:967.
21. Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol 2009;19:12.
24. Racker E. Why do tumor cells have a high aerobic glycolysis? J Cell Physiol 1976;89:697.
25. Sonveaux P, Vegran F, Schroeder T, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 2008;118:3930.
27. Guppy M, Leedman P, Zu X, et al. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J 2002;364:309.
29. DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007;104:19345.
32. Hatzivassiliou G, Zhao F, Bauer DE, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005;8:311.
35. Lockart RZ Jr, Eagle H. Requirements for growth of single human cells. Science 1959;129:252.
37. Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev 2009;89:381.
40. Hawkins RA, Phelps ME. PET in clinical oncology. Cancer Metastasis Rev 1988;7:119.
41. Ben-Haim S, Ell P. 18F-FDG PET and PET/CT in the evaluation of cancer treatment response. J Nucl Med 2009; 50:88.
46. Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer 2008;8:865.
47. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008;30:393.
48. Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 2008;15:678.
49. Majumder PK, Febbo PG, Bikoff R, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med 2004;10:594.
50. Dang CV, Kim JW, Gao P, et al. The interplay between MYC and HIF in cancer. Nat Rev Cancer 2008;8:51.
57. Flier JS, Mueckler MM, Usher P, et al. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987;235:1492.
58. Birnbaum MJ, Haspel HC, Rosen OM. Transformation of rat fibroblasts by FSV rapidly increases glucose transporter gene transcription. Science 1987;235:1495.
60. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006;7:606.
71. Cheung EC, Vousden KH. The role of p53 in glucose metabolism. Curr Opin Cell Biol 2010;22:186.
75. Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science 2006;312:1650.
79. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2007;8:774.
82. Tee AR, Blenis J. mTOR, translational control and human disease. Semin Cell Dev Biol 2005;16:29.
83. Dann SG, Thomas G. The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett 2006;580:2821.
85. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006;441:424.
86. Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol 2008;3:427.
87. Jin S, White E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy 2008;4:563.
88. Wang RC, Levine B. Autophagy in cellular growth control. FEBS Lett 2010;584:1417.
90. Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000;287:848.
95. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 2006;25:4675.
98. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321:1807.
100. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058.
101. Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med 2009;360:813.
110. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462:739.
116. Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 1948;238:787.
118. Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005;330:1304.
127. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 2008;8:915.
128. Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature 2009;458:725.
132. Laszlo J, Humphreys SR, Goldin A. Effects of glucose analogues (2-deoxy-D-glucose, 2-deoxy-D-galactose) on experimental tumors. J Natl Cancer Inst 1960;24:267.
141. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 2008;13:472.
148. Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2:31.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


