Cancer and cell death
John C. Reed, MD, PhD
Overview
Cell death is a normal facet of human physiology, ensuring tissue homeostasis by offsetting cell production with cell demise. Neoplasms arise in part because of defects in physiological cell death mechanisms, contributing to pathological cell expansion when genetic or epigenetic alternations impart a selective survival advantage to premalignant or malignant cells. Defects in normal cell death pathways also contribute to cancer progression by permitting progressively aberrant cell behaviors, while also desensitizing tumor cells to immune-mediated attack, radiation, and chemotherapy. Multiple mechanisms that account for dysregulation of cell death mechanisms in human malignancies have been identified, providing insights into cancer pathogenesis and suggesting targets for therapeutic intervention based on the concept of restoring sensitivity to natural pathways for triggering cell suicide.
Introduction
Evasion of endogenous cell death processes represents one of the cardinal characteristics of cancer.1 Enormous amounts of cell death occur on a daily basis within the human body, estimated at 50–70 billion cells per day for the average adult. In bulk terms, this amounts to a mass of cells equal to an entire body weight annually. This “programmed” cell death offsets daily cell production resulting from cell division, achieving tissue homeostasis. Cell death can proceed by several distinct mechanisms. Apoptosis and necrosis are the two broadest categories of cell death mechanisms, but specialized forms of cell death have also been described, including necroptosis, pyroptosis, ferrotopsis, and autophagic cell death. Apoptosis is the most common nonpathological route of cell demise in the context of normal tissue homeostasis and mammalian development.2–4
During recent years, numerous examples have been delineated whereby cancer cells place roadblocks in the way of endogenous cell death mechanisms, thus endowing neoplastic cells with a selective survival advantage. Several molecules that create barriers to cell death within tumors have been identified, providing insights into pathogenic mechanisms of human neoplasia and cancer—in addition to suggesting targets for drug discovery, with the aim of restoring the integrity of natural pathways for cell suicide and thereby stimulating auto-destruction of cancer cells.5, 6 Alternatively, malignant cells can escape demise by silencing or neutralizing endogenous activators of cell death, with several examples of such mechanisms already elucidated. Replacing or reawakening these endogenous stimulators of programmed cell death (PCD) defines another emerging strategy for potentially eradicating tumors.7, 8
Defects in the natural mechanisms regulating cell death aid cancers in many ways.2, 5, 6 First, activation of oncogenes such as C-MYC drives proliferation of transformed cells and also promotes apoptosis, unless counteracted by cell survival proteins—thus constituting a type of oncogene “complementation.”9 A second, closely related, benefit to tumors of defective cell death is that various cell cycle “checkpoints” trigger apoptosis of cells undergoing aberrant cell replication or division. Thus, defects in apoptosis and other cell death mechanisms can permit aberrant cell division. Third, genomic instability is also aided by defects in cell death mechanisms because mutations introduced by DNA replication errors and chromosome segregation errors would normally trigger cell suicide. Fourth, defects in cell death mechanisms allow growth factor (or hormone)-independent cell survival, thus helping transformed cells to escape normal paracrine and endocrine growth control mechanisms. Fifth, cell death induced by hypoxia and metabolic stress, conditions that occur in the microenvironments of most rapidly growing solid tumors, are neutralized by defects in cell death machinery that occur in cancers. Sixth, invasion and metastasis of solid tumors may also be enabled by defects in cell death mechanisms, given that epithelial cells normally undergo apoptosis when they lose their attachment to extracellular matrix through integrins.10 Seventh, defects in cell death mechanisms contribute to avoidance of immune surveillance mechanisms by making it more difficult for cytolytic T-cells (CTLs) and natural killer (NK) cells to kill tumor cells.
In this chapter, the major pathways for cell death are reviewed and examples of their defects in cancers are described, including hypoexpression (hypoactivity) of pro-death genes and overexpression (hyperactivity) of cytoprotective genes. Insights into the mechanisms that lead to cell death dysregulation in cancers have also sparked a variety of oncology drug discovery and development efforts, which are also described briefly. With apology to the many contributors to the field of cell death biology, only representative references are cited for purposes of brevity.
Pathways for cell death
Several endogenous pathways for triggering cell suicide have been delineated and a variety of ways for cataloging these cell death mechanisms have been utilized by the research community.11, 12 A general construct for categorizing cell death mechanisms can be attributed to the essential involvement of a family of intracellular proteases called “Caspases” (cysteine-dependent aspartate-specific proteases).13 Broadly, cell death mechanisms can thus be divided into Caspase-dependent versus Caspase-independent, though mixtures of these two basic mechanisms are commonly observed in various settings.
Caspase-dependent cell death
Caspase-dependent cell death culminates in the constellation of morphological changes meeting criteria for “apoptosis,” where cells detach from extracellular matrix, become rounded and shrunken in volume, with nuclear chromatin condensation, nuclear fragmentation, and the membrane “bubbling” with membrane protrusions (blebs) that can bud off to create apoptotic bodies. These morphological changes can be attributed directly or indirectly to the multitude of proteolytic cleavage events mediated by Caspases, a full accounting of which is still under investigation using comprehensive proteomics methods.14
Caspases operate in hierarchical networks of upstream initiators and downstream effectors, where they cleave and activate each other to create amplifiable cascades of proteolysis.15, 16 These proteases are present as inactive zymogens within the cytosol of essentially all animal cells. Activation of the most proximal Caspases typically occurs through protein interactions that encourage clustering of the inactive zymogens.16 Once activated, these upstream initiators then cleave and activate downstream effector Caspases. The upstream initiator Caspases contain N-terminal prodomains that mediate protein interactions with other proteins involved in Caspase regulation—namely, CARDs (Caspase-associated recruitment domains) and DEDs (death effector domains) (Table 1). Genomic mutations that inactivate Caspases have been described in a variety of cancers, though overall they are relatively rare.
Table 1 Domains associated with cell death proteins
| Domain | Proteins (examples) |
| Caspase catalytic domain | Caspase family cysteine proteases |
| Caspase-associated recruitment domain (CARD) | Caspases-1, -4, -5, -9; Apaf1; ASC; NLRP1, 4 |
| Death domain (DD) | TNFR1, FAS, DR4, DR5, TRADD, RAIDD, RIP1 |
| Death effector domain (DED) | Caspases-8, -10; FADD; c-FLIP |
| Baculovirus IAP repeat (BIR) | XIAP, c-IAP1, c-IAP2, Livin, Apollon, ML-IAP, NAIP |
| Bcl-2 homology (BH) | Bd-2, Bcl-XL, Mcl-1, Bax, Bak, Bim, Bid |
Multiple pathways that lead to Caspase activation and that thereby cause apoptosis or variations of apoptosis have been delineated (Figure 1). For example, CTLs and NK cells introduce apoptosis-inducing proteases, particularly Granzyme B (a serine protease), into target cells via perforin-mediated pores.17 Enzymologically, Granzyme B is a serine rather than a cysteine protease. However, similar to the Caspases, Granzyme B cleaves its substrates at Asp residues.18 Granzyme B is capable of cleaving and activating multiple Caspases. Tumors overcome this pathway for cell death by expressing various immune ligands for checkpoint receptors expressed on T cells (PD1, Tim3, LAG3, etc.) that suppress T-cell activation or by expressing ligands for inhibitory NK receptors (killer immunoglobulin-like receptors (KIRs)).
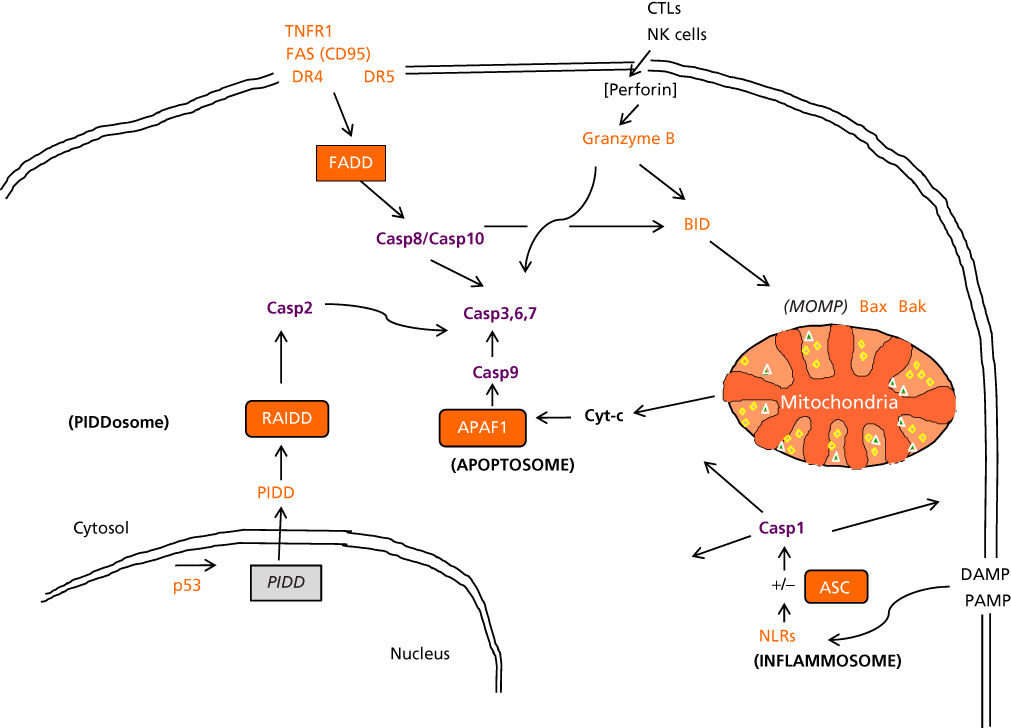
Figure 1 Caspase activation pathways. The major pathways for Caspase activation in mammalian cells are presented. The schematic represents an oversimplification of the events that occur in vivo. The extrinsic (left, upper) is induced by members of the TNF- family of cytokine receptors such as TNFR1, Fas, and the TRAIL Receptors, DR4 and DR5. These proteins recruit adapter proteins to their cytosolic DDs, including FADD, to assemble “death-inducing signaling complexes” (DISCs), which bind DED-containing pro-Caspases, particularly pro-Caspase-8, inducing their activation. Caspase-8 (and Caspase-10 in humans) cleaves downstream effector proteases, such as Caspases-3, -6, and-7, as well as pro-apoptotic Bcl-2 family member Bid, which is an agonist of MOMP-inducing Bax and Bak. CTLs and NK cells introduce the protease Granzyme B into target cells (right, upper). This protease cleaves and activates multiple members of the Caspase-family, as well as Bid. The intrinsic pathway (left, lower) is initiated by release of Cyt-c from mitochondria, induced by various stimuli, including elevations in the levels of pore-forming pro-apoptotic Bcl-2 family proteins such as Bax and Bak. In the cytosol, Cyt-c binds and activates Apaf-1, allowing it to associated with and activate pro-Caspase-9, forming “apoptosomes.” Active Caspase-9 directly cleaves and activates the effector proteases, Caspase-3 and -7. Exogenous pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs) activate NLR family proteins to cause their oligomerization and assembly of “inflammasomes” that may contain PYD-containing adapter protein ASC in many cases, which binds pro-Caspase-1. Activated Caspase-1 cleaves a variety of cellular proteins, including cytokines as well as plasma membrane proteins that promote osmotic stress and cell death.
Another Caspase-activation pathway is stimulated by tumor necrosis factor (TNF)-family receptors. Eight of the ∼30 known members of the TNF family in humans contain a so-called death domain (DD) in their cytosolic tails.19, 20 Several of these DD-containing TNF-family receptors use Caspase activation as a signaling mechanism, including TNF receptor-1 (TNFR1)/CD120a; Fas/APO1/CD95; death receptor-3 (DR3)/Apo2/Weasle; DR4/TrailR1; DR5/TrailR2; and DR6. Ligation of these receptors at the cell surface results in receptor clustering and recruitment of several intracellular proteins, including certain pro-Caspases, to the cytosolic domains of these receptors, forming a “death-inducing signaling complex” (DISC) that triggers Caspase activation and leads to apoptosis.21 The specific Caspases recruited to the DISC are Caspase-8 and, in some cases, Caspase-10. These Caspases contain DEDs in their N-terminal prodomains that bind to a corresponding DED in FADD, a bipartite adapter protein containing a DD and a DED. FADD functions as a molecular bridge between the DD and DED domain families, and is in fact the only protein in the human genome with this dual domain structure. Consequently, cells from mice in which the fadd gene has been knocked out are resistant to apoptosis induction by TNF-family cytokines and their receptors. Cells derived from Caspase-8 knockout mice also fail to undergo apoptosis in response to ligands or antibodies that activate TNF-family death receptors, demonstrating an essential role for this Caspase in this pathway.22, 23 However, mice lack the highly homologous protease, Caspase-10, which is found in humans, having arisen from an apparent gene duplication on chromosome 2.15 Thus, Caspases-8 and -10 may play redundant roles in human cells. Genomic mutations in genes encoding TNF-family ligands or receptors have been documented in some types of cancer. For example, somatic mutations in the FAS (CD95) gene have been found in lymphoid malignancies.24, 25 Missense mutations within the DD of Fas (CD95) are associated with retention of the wild-type allele, suggesting a dominant-negative mechanism, whereas missense mutations outside the DD are associated with allelic loss.
Mitochondria also play important roles in apoptosis, releasing Cytochrome-c (Cyt-c) into the cytosol, which then causes assembly of a multiprotein Caspase-activating complex, referred to as the apoptosome.26–29 The central component of the apoptosome is Apaf1, a Caspase-activating protein that oligomerizes upon binding Cyt-c and which specifically binds pro-Caspase-9. Apaf1 and pro-Caspase-9 interact with each other via their CARDs. Such CARD–CARD interactions play important roles in many steps in apoptosis pathways. The central importance of the Cyt-c-dependent pathway for apoptosis is underscored by the observation that cells derived from mice in which either the apaf1 or pro-Caspase-9 genes have been ablated are incapable of undergoing apoptosis in response to agents that trigger Cyt-c release from mitochondria.30, 31 Nevertheless, such cells can die by non-apoptotic routes,32 demonstrating that mitochondria control both Caspase-dependent and Caspase-independent cell death pathways (see below). Mitochondria can also participate in cell death pathways induced via TNF-family death receptors, through cross-talk mechanisms involving proteins such as Bid, which become activated upon proteolytic cleavage by Caspase-8 and then stimulate Cyt-c release from mitochondria.33 However, mitochondrial (“intrinsic”) and death receptor (“extrinsic”) pathways for Caspase activation are fully capable of independent operation in most types of cells.34 Tumor cells can evolve a variety of mechanisms for averting mitochondria-dependent apoptosis, as outlined below.
Activation of Caspase-1 in the context of infection and inflammation has been shown to stimulate a Caspase-dependent form of cell death called pyroptosis.35 Caspase-1 has a CARD-containing prodomain that can bind directly to the CARDs of NLR (nucleotide-binding domain and leucine-rich repeat domain) proteins, such as NLRP1 (NALP1). Alternatively, the CARD of Caspase-1 can bind the CARD of a bipartite adapter protein ASC (Pycard), which also contains a PRYIN domain (PYD) that binds PYDs of NLR family proteins such as NLRP3 (NALP3) or proteins such as AIM2. NLRs oligomerize in response to binding components of pathogens or molecules elaborated during tissue injury, forming “inflammasomes” that recruit and activate Caspase-1.36–38 The relevance of pyrotopsis to cancer biology is largely unknown.
Caspase-2 is another CARD-carrying member of the Caspase family. Its CARD interacts specifically with the CARD of the bipartite adapter protein RAIDD, which additionally carries a DD.39 Caspase-2 activation has been linked to genotoxic stress via a pathway whereby tumor suppressor p53 induces expression of PIDD, a DD-containing protein that binds the DD of RAIDD, oligomerizes, and assembles into a multiprotein complex termed the “PIDDosome”.40
Additional Caspase activation mechanisms have been elaborated, though it is less clear how prominently they figure in the pathophysiology of cell death.41–44
Caspase-independent cell death
Multiple Caspase-independent cell death mechanisms have been identified, with a few prominent examples mentioned here (Figure 2). Necrosis can be initiated by many stimuli. It occurs when cells are incapable of maintaining osmotic equilibrium such that plasma membrane integrity is compromised, resulting in cell swelling and rupture. Organelles within cells during necrosis also typically swell and rupture, including mitochondria and lysosomes, releasing molecules that promote cell death. Initiators of necrosis include circumstances that compromise bioenergetics such as hypoxia and hypoglycemia, resulting in inadequate adenosine triphosphate (ATP) concentrations for powering plasma membrane ionic pumps, as well as insults to the plasma membrane such as serum complement factors that create pores. Necrosis is relevant to cancer biology in the context of rapidly growing tumors that outstrip their vascular supply (insufficient angiogenesis), creating regions of hypoxia and nutrient insufficiency.
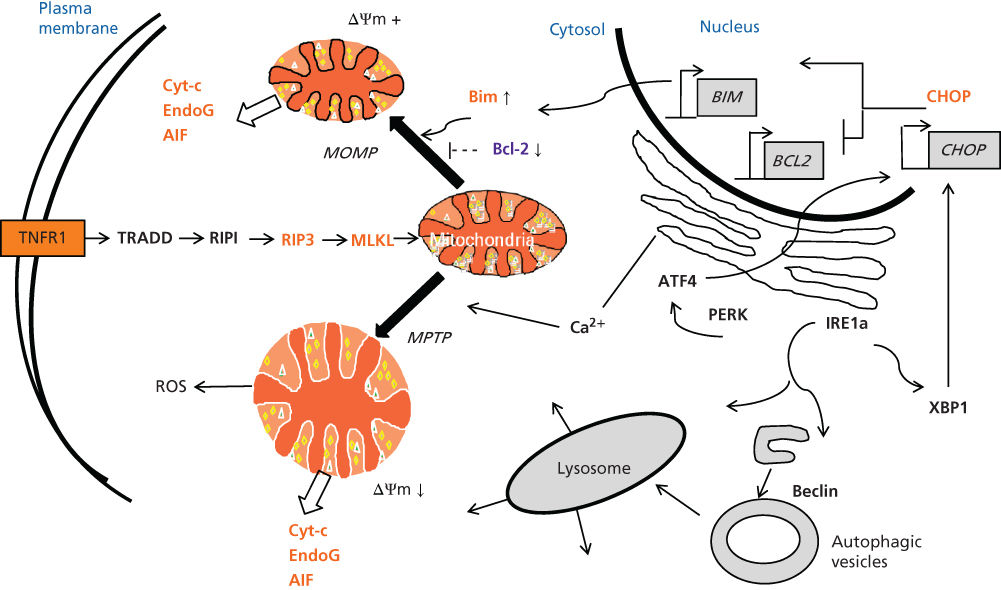
Figure 2 Caspase-independent cell death pathways. The TNFR1-mediated pathway for necrosis (necroptosis) involves a cascade of events that include recruitment of Rip1, which in turn activates Rip3, which activates MLKL and which causes mitochondrial and probably lysosomal changes that stimulate ROS generation and lead to necrosis. The cytosolic DD of TNFR1 binds the DD-containing adapter protein TRADD, which binds the DD of Rip1. The kinase cascade involves Rip1, followed by Rip3, followed by MLKL. Mitochondria release proteins stored in the intermembrane space, including Cyt-c, Endonuclease G, and AIF. Release can be induced by MOMP (upper), which is regulated by Bcl-2 family proteins and which does not involve organelle swelling. Alternatively, release of mitochondrial can occur as a result of by MPTP complex opening, which leads to organelle swelling and loss of the electrochemical gradient (ΔΨm). MPTP opening is induced by Ca2+ influx and oxidative stress, among other stimuli. Accumulation of unfolded proteins in the ER results in UPR signaling, including activation of IRE1α, which splices XBP1 mRNA to generate XBP1 protein (transcription factor) and activation of the kinase PERK, which stimulates phosphorylation events that promote ATF4 mRNA translation and production of transcription factor ATF4. The UPR transcription factors (XBP1, ATF4, and (not shown) ATF6) converge on the promoter of the CHOP gene, which encodes a transcription factor that upregulates pro-apoptotic gene BIM and downregulates anti-apoptotic gene BCL-2. Autophagic vesicles emerge from the ER, form double membrane vesicles, which fuse with lysosomes (bottom, right).
Mitochondria-initiated cell death pathways can also lead to Caspase-independent cell death, in addition to the aforementioned Caspase-dependent pathway downstream of Cyt-c.45 In addition to Cyt-c, mitochondria also release several other proteins of relevance to apoptosis, including Endonuclease G, AIF (an activator of nuclear endonucleases), and Smac (Diablo) and Omi (HtrA2), antagonists of a family of Caspase-inhibitory proteins known as the inhibitor of apoptosis proteins (IAP)s (see below).46, 47 Moreover, distinguishing mitochondria-driven apoptotic from non-apoptotic cell death can be challenging in many contexts due to the similar morphological features caused probably by some of the proteins released from these organelle such as EndoG and AIF, which promote chromatin condensation and DNA fragmentation. The mitochondrial mechanisms for apoptotic and non-apoptotic cell death are activated by myriad stimuli, including growth factor deprivation, oxidants, Ca2+ overload, DNA-damaging agents, microtubule-modifying drugs, and much more. In this sense, mitochondria are sometimes viewed as central integrators of cell stress signals that ultimately dictate cell life and death decisions.48–50
Uniformly, these mitochondrial cell death mechanisms involve compromises of mitochondrial membrane integrity, but the membrane permeability change can occur via different mechanisms.51 In the context of insults such as ischemia-reperfusion injury (where oxidative stress occurs due to reactive oxygen species (ROS) and nitrosyl free radicals) and Ca2+ dysregulation where cytosolic free Ca2+ levels rise (due to loss of plasma membrane integrity or escape from the endoplasmic reticulum [ER]), osmotic homeostasis of mitochondria is perturbed due to opening of the mitochondrial permeability transition pore (MPTP) complex that spans inner and outer membranes at contact sites where they come into close apposition.52–54 The change in permeability in the inner membrane of mitochondria allows water and electrolytes to enter the protein-rich matrix of mitochondria, resulting in swelling of the organelle and eventually popping of the outer membrane, whose surface area is smaller than the extensively folded inner membrane with its cristae. During pathological opening of the MPTP complex, the electrochemical gradient (proton gradient) that is essential for ATP production is lost, and thus bioenergetics is severely compromised. An essential component of the MPTP complex is the adenine nucleotide translocator (ANT), which is thought to be chiefly responsible for the permeability transition that leads to mitochondrial swelling.
Alternatively, cell death mechanisms can involve the phenomenon of mitochondrial outer membrane permeabilization (MOMP), where the outer membrane becomes selectively permeabilized, allowing release of Cyt-c and other proteins that reside in the intermembrane space.51, 55, 56 With MOMP, the electrochemical gradient is preserved and mitochondrial osmotic homeostasis is maintained. MOMP is caused by pro-apoptotic members of the Bcl-2 family (see below), namely Bax and Bak (possibly also Bok).57–60 Though others may exist, an established mechanism for triggering MOMP entails interaction of pro-apoptotic proteins such as Bid, Bim, and Puma with Bax and/or Bak, causing oligomerization of Bax and Bak in the mitochondrial outer membrane and the formation of ill-defined pores that permit escape of Cyt-c and other proteins. MOMP induced by Bax and/or Bak thus initiates the mitochondrial pathway for Caspase-dependent cell death and apoptosis by releasing Cyt-c, but it also secondarily causes Caspase-independent necrotic cell death by uncoupling of oxidative phosphorylation (when Cyt-c becomes limiting) and causing diversion of electrons from the respiratory chain into production of toxic free radicals.61
Given the diversity of cell death mechanisms initiated by mitochondria, it is not surprising that tumors evolve a variety of alternations in these organelles and their regulation. The metabolic changes that convert the bioenergetic signature of tumor cells from dependence on aerobic respiration to non-aerobic glycolysis are well known (Warburg effect), and could be argued to make tumor cells less dependent on mitochondria. Indeed, some tumors seem to have a diminution in the numbers of mitochondria compared to normal cells of the same cell lineage.62
Cell death induced by CTCs and NK cells can also occur via Caspase-independent mechanism. In this regard, Granzyme B directly cleaves some of the same substrates as Caspases,63, 64 thereby potentially bypassing the need for Caspases to cause proteolytic events associated with apoptosis. Also, the cytotoxic granules of CTLs and NK cells contain several other death-promoting molecules besides Granzyme B.17 Moreover, the perforin-mediated pores created in the plasma membrane of target cells by CTLs and NK cells can also lead to osmotic disequilibrium and thus Caspase-independent cell death. The diversity of cytolytic mechanisms invoked by CTLs and NK cells bodes well for attempts to harness the immune system as a weapon for attacking cancers, since specific blocks to cell death in tumors (see below) might be overwhelmed by the multiple parallel cytotoxic mechanisms initiated by these immune cells.
Necroptosis is the term that has been applied to a Caspase-independent cell death mechanism initiated by TNFR1. When Caspase-8 is incapacitated, TNFR1 can stimulate a non-apoptotic cell death signaling pathway that involves the serine/threonine protein kinases Rip1, Rip3, and MLKL (mixed lineage kinase domain-like).65–67 Rip3-dependent necroptosis occurs through a process involving ROS generated by mitochondria. Necroptosis was initially identified by studies of tumor cells where TNFα was shown to kill via a Caspase-independent mechanism, suggesting that this cell death pathway could provide a conduit for attacking cancers.
Both Caspase-dependent and Caspase-independent cell death signaling mechanisms have been implicated in endoplasmic reticulum stress (ER stress). A wide variety of microenvironmental conditions (including those relevant to the tumor microenvironment) can cause the accumulation of unfolded proteins in the ER, triggering an adaptive signal transduction response called the unfolded protein response (UPR).68–70 Among these UPR events is increased transcription of a gene encoding the pro-apoptotic transcription factor CHOP, which in turn stimulates expression of the gene encoding DR5 (death receptor 5; TRAIL Receptor-2), causing Caspase-8-dependent apoptosis. In addition, CHOP has been reported to directly stimulate transcription of the gene encoding Bim, a pro-apoptotic member of the Bcl-2 family (see below) that stimulates Bax/Bak oligomerization in mitochondrial membranes to induce MOMP and release Cyt-c and other proteins from mitochondria. Thus, ER stress has multiple potential routes of stimulating both Caspase-dependent and Caspase-independent cell death, with the predominant pathway probably varying among cell types and pathophysiological contexts.
Furthermore, the important role that the ER plays in regulating intracellular Ca2+ homeostasis provides another potential link to cell death mechanism where changes in cytosolic free Ca2+ contribute (reviewed in70–72). When extreme, liberation of Ca2+ from the lumen of the ER can trigger a variety of downstream signaling events that promote cell death. Also, the ER collaborates with mitochondria in many ways that can either promote cell life or cause death. ER membranes from close contacts with mitochondria to create structures where Ca2+ effluxes from ER into mitochondria, aiding bioenergetics. However, too much Ca2+ entry into mitochondria triggers opening of the MPTP complex, such that mitochondria swell and eventually rupture.
Autophagy (“self-eating”) is a catabolic mechanism where senescent proteins and organelles become entrapped in double membrane vesicles that bud off from the ER and that are transported to lysosomes for degradation of their contents. The physiological roles of autophagy include protein homeostasis, supplementing the ubiquitin-proteasome system for removal of unfolded, defective, and aged proteins, as well as providing a source of substrates for maintaining metabolism during times of nutrient deprivation.73, 74 In the context of nutrient deprivation, autophagy can contribute to cell survival and is thought to aid tumors at some points in their evolution. However, when taken to an extreme, mechanisms that rely on components of the autophagy machinery have been reported to cause cell death via a process called autophagic cell death, which is Caspase independent.75 Under some conditions, exposure of tumor cells to cytotoxic chemotherapeutic drugs can trigger autophagic cell death.76
Additional Caspase-independent cell death mechanisms have been described, including lysosome-dependent cell death,77 iron-dependent cell death (ferroptosis),78 and others,11 which are not reviewed here. All of them may be relevant to cancer biology at some level in some contexts.
Cell death resistance mechanisms used by cancers
Cancer cells have been shown to put the brakes on Caspases and thereby suppress apoptosis by at least three fundamental mechanisms—(1) preventing activation of Caspase zymogens (pro-enzymes); (2) neutralizing active Caspases (active enzymes); and (3) suppressing expression of genes encoding Caspases or Caspase-activating proteins (see below). Malignant cells also develop a variety of ways of counteracting Caspase-independent cell death mechanisms. Some of the more prominent examples understood to date are outlined here.
IAPs
Endogenous suppressors of Caspases include IAPs (n = 8 in humans),15, 79, 80 an evolutionarily conserved family of proteins that directly bind active Caspases and that either suppress their protease activity or that target active Caspases for destruction.79, 81, 82 Overexpression of IAPs occurs in cancers and leukemias, making it more difficult to propagate the Caspase-dependent proteolytic events necessary for apoptotic cell death.
IAPs are characterized by the presence of protein interaction domains called BIRs (baculovirus internal repeats), numbering between 1 and 3 per protein83 (Table 1). Most IAPs also carry RING domains that endow them with E3 ligase activity through interactions with ubiquitin-conjugating enzymes (UBCs). Some of the apoptogenic proteins released from mitochondria, notably SMAC and HtrA2,84, 85 bind certain BIRs and thereby compete for protein interactions on the surface of IAPs. In many cases, Smac binding to IAPs induces their polyubiquitination and proteasomal degradation. Thus, factors that cause MOMP take the brakes off the Caspases by eliminating various IAP family proteins.
The most established Caspase inhibitor among the IAP family members is XIAP (so called because its encoding gene resides on the X-chromosome). The XIAP protein contains 3 BIR domains. BIR2 of XIAP (and an adjacent segment) binds downstream effector proteases, Caspases-3 and -7, to potently suppress apoptosis at a distal point. BIR3 of XIAP binds upstream initiator protease, Caspase-9, to suppress an apical step in the mitochondrial pathway for apoptosis. Overexpression of XIAP mRNA and protein has been reported in cancers and leukemias, apparently caused by epigenetic mechanisms rather than genomic lesions to the XIAP gene. XIAP protein stability may also be increased in tumors with Akt hyperactivity.86
The c-IAP-1 (BIRC2) and c-IAP2 (BIRC3) proteins are also capable of binding to Caspases-3, -7, and -9, though they are far less potent as direct enzymatic inhibitors and may rely on their E3 ligase activity for controlling Caspase degradation. However, these IAP family members also participate in other cell-death-relevant mechanisms by impacting signal transduction by TNF-family receptors.65, 66, 87 Binding of cytokine TNFα to TNFR1 can trigger at least three different signaling pathways, each involving overlapping but distinct protein complexes that are assembled at the receptor.88 In addition to the pathways for Caspase-dependent and Caspase-independent cell death outlined above, TNFR1 also stimulates a cell survival pathway in which c-IAP1 and c-IAP2 participate. In this regard, the BIR3 domains of c-IAP1 and c-IAP2 directly bind the kinase Rip1. In the TNFR1-mediated survival pathway, Rip1 forms protein complexes that contain the E3 ligases c-IAP1, c-IAP2, and TRAF2, stimulating noncanonical (lysine 63, rather than lysine 48) ubiquitination of Rip1 to initiate a signal transduction pathway that causes activation of transcription factor NF-κB.89, 90 NF-κB influences the expression of many target genes involved in host defenses and immune regulation, among which are several genes that suppress apoptosis91 (see below). As a result, this NF-κB pathway nullifies the Caspase pathway, negating apoptosis, in addition to accounting for the untoward inflammatory actions of this cytokine. In addition, the Rip3-dependent pathway for necrotoposis is suppressed by c-IAP1 and c-IAP2, probably via their roles as E3 ligases and possibly involving ubiquitin-/proteasome-mediated reductions in Rip3 protein levels. In human cancers, chromosomal translocations that deregulate c-IAP2 by creating a chimeric protein mucosa-associated lymphoid tissue (MALT-c-IAP2) have been described in lymphomas.92, 93 In addition, the genomes of some cancers have amplification of the gene encoding TRAF2, the c-IAP1/c-IAP2-binding protein implicated in TNF-family receptor signaling. Overexpression of c-IAP1 and c-IAP2 mRNAs and proteins, presumably via epigenetic mechanisms, is also reported. In this regard, c-IAP2 is among the direct transcriptional targets of NF-κB, and hyperactivity of the canonical (RelA) and alternative (RelB) pathways is well established in human malignancies of various types.
Additional members of the IAP family, not described here (Survivin, Apollon/Bruce, ML-IAP, etc.), also have interesting mechanisms of interacting with components of cell death pathways; and they can have other roles beyond cell death regulation. For example, the Survivin protein plays a fundamental role in chromosome segregation and cytokinesis.94
c-FLIP
The c-FLIP protein provides another example of an endogenous modulator of Caspases whose overexpression is a common occurrence in tumors.95, 96 FLIP is structurally similar to certain Caspases, containing a pair of DEDs analogous to Caspases-8 and -10. It directly binds and suppresses activation of Caspases-8 and -10 in the context of TNF-family death receptor signaling. However, the role of c-FLIP is more complex than merely suppressing Caspase-8 or -10 activation, as it also collaborates with these Caspases in poorly understood ways (probably involving Caspase-8, MALT, and Bcl-10) to promote NF-κB activation.97 Testimony to this complex interaction is also found in the observation that knockout of the genes encoding either c-Flip or Caspase-8 in mice results in similar embryonic lethal phenotypes. In addition to promoting NF-κB activation, the c-FLIP gene is itself also a direct transcriptional target of NF-κB, whose expression is strikingly upregulated (positive feedback).98
Bcl-2
Among the central regulators of MOMP are members of the Bcl-2 family, evolutionarily conserved proteins that either promote (e.g., Bax; Bak) or suppress (e.g., Bcl-2, Bcl-XL) MOMP.99–101 The Bcl-2-family proteins (n = 26 in humans)15 physically interact with each other, forming complex networks of homo- and heterodimers that steer the cell toward either life or death.102, 103 Numerous examples of altered expression and function of Bcl-2-family proteins have been identified in cancers, accounting for intrinsic resistance to cell death induced by myriad stimuli, including most cytotoxic anticancer drugs and x-irradiation, growth factor deprivation and therapeutic inhibitors of growth factor receptor signal transduction pathways.
Many members of the Bcl-2-family have a hydrophobic stretch of amino acids near their carboxyl-terminus that anchors them in the outer mitochondrial membrane. In contrast, other Bcl-2-family members such as Bid, Bim, and Bad lack these membrane-anchoring domains, but dynamically target to mitochondria in response to specific stimuli. Still others have the membrane-anchoring domain but keep it latched against the body of the protein, until stimulated to expose it (e.g., Bax).
Based on their predicted (or experimentally determined) three-dimensional structures, Bcl-2 family proteins can be broadly divided into two groups. One subset of these proteins is probably similar in structure to the pore-forming domains of bacterial toxins, such as the colicins and diphtheria toxin.104–106 These α-helical pore-like proteins include both anti-apoptotic proteins (Bcl-2, Bcl-XL, Mcl-1, Bfl-1, Bcl-W, Bcl-B), as well as pro-apoptotic proteins (Bax, Bak, Bok, and Bid). Most of the proteins in this subcategory can be recognized by conserved stretches of amino acid sequence homology, including the presence of Bcl-2 homology (BH) domains, BH1, BH2, BH3, and sometimes BH4 (Table 1). However, this is not uniformly the case, as the Bid protein contains only a BH3 domain but has been determined to share the same overall protein-fold with Bcl-XL, Bcl-2, and Bax. Where tested to date, these proteins have all been shown to form ion-conducting channels in synthetic membranes in vitro, including Bcl-2, Bcl-XL, Bax, and Bid,107 but the significance of this pore activity remains largely unclear. The other subset of Bcl-2 family proteins appears to have in common only the presence of the BH3 domain, including Bad, Bik, Bim, Hrk, Bcl-GS, p193, APR (Noxa), and PUMA. These “BH3-only” proteins are uniformly pro-apoptotic. Their cell-death-inducing activity depends, in most cases, on their ability to dimerize with anti-apoptotic Bcl-2 family members, functioning as trans-dominant inhibitors of proteins such as Bcl-2 and Bcl-XL.101, 102 However, a select group of them (Bim, Bid, Puma) interact with pro-apoptotic proteins Bax and Bak, functioning as agonists that induce their oligomerization in mitochondrial membranes to stimulate MOMP.
The BH3 domains mediate dimerization among Bcl-2 family proteins (Figure 3
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


