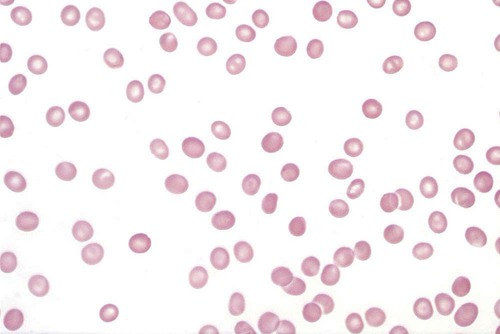
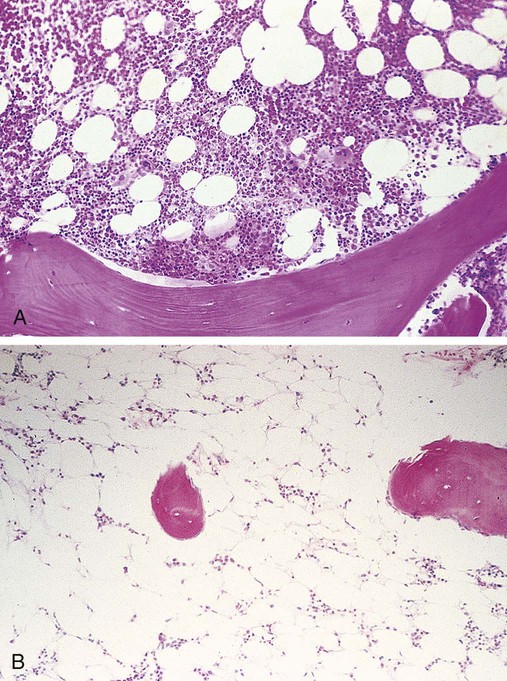
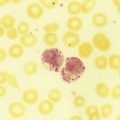
After completion of this chapter, the reader will be able to: 1. Describe the clinical consequences of bone marrow failure. 2. Describe the etiology of acquired and inherited aplastic anemias. 3. Discuss the pathophysiologic mechanisms of acquired and inherited aplastic anemias. 4. Describe the characteristic peripheral blood and bone marrow features in aplastic anemia. 5. Classify aplastic anemia as nonsevere, severe, or very severe based on laboratory tests. 6. Discuss treatment modalities for acquired and inherited aplastic anemia and the patients for whom each is most appropriate. 7. Differentiate among causes of pancytopenia based on laboratory tests and clinical findings. 8. Discuss the possible relationship between defects in the telomerase complex and bone marrow failure in acquired and inherited aplastic anemia. 9. Compare and contrast the pathophysiology, clinical picture, and laboratory findings in transient erythroblastopenia of childhood, Diamond-Blackfan anemia, and congenital dyserythropoietic anemia. 10. Describe the mechanisms causing cytopenia in myelophthisic anemia and anemia of chronic kidney disease. 1. What term is used to describe a decrease in all cell lines in the peripheral blood? 2. Which anemia of bone marrow failure should be considered? 3. How would an increase in either reticulin or blasts alter the preliminary diagnosis? 4. How would the severity of this patient’s condition be classified? 5. What treatment modality would be considered for this patient? Aplastic anemia is a rare but potentially fatal bone marrow failure syndrome. In 1888, Ehrlich provided the first case report of aplastic anemia involving a patient with severe anemia, neutropenia, and a yellow hypocellular marrow on postmortem examination.1 The name aplastic anemia was given to the disease by Vaquez and Aubertin in 1904.2 The characteristic features of aplastic anemia include pancytopenia, reticulocytopenia, bone marrow hypocellularity, and depletion of hematopoietic stem cells (Box 21-1). Approximately 80% to 85% of cases of aplastic anemias are acquired, and 15% to 20% are inherited.3 Box 21-2 provides an etiologic classification.3–5 Acquired aplastic anemia is classified as idiopathic when the cause is unknown and as secondary when the cause can be identified. Approximately 70% of cases are idiopathic and 10% to 15% are secondary.3 Idiopathic and secondary aplastic anemia have similar clinical and laboratory findings. Initially, patients may manifest a macrocytic or normocytic anemia with reticulocytopenia. Depending on the progression of the bone marrow failure, pancytopenia may develop slowly or progress at a rapid rate with complete cessation of hematopoiesis. In North America and Europe, the incidence per year approximates 2 in 1 million.6 In Asia and East Asia, the incidence is two to three times higher than in North America or Europe, which may be due to environmental or genetic differences.7 Aplastic anemia can occur at any age, with the highest frequency at 15 to 25 years and the second highest frequency over the age of 60.4,6,8 There is no gender difference in incidence.6 The cause of the bone marrow failure in idiopathic aplastic anemia is unknown. Secondary aplastic anemia is associated with exposure to certain drugs, chemicals, radiation, or infectious agents. Cytotoxic drugs, radiation, and benzene suppress the bone marrow in a predictable, dose-dependent manner.4,5 Depending on the dose and exposure time, the bone marrow generally recovers after withdrawal of the agent. About 90% of cases of secondary aplastic anemia occur due to idiosyncratic reactions to drugs or chemicals. In idiosyncratic reactions, the bone marrow failure is unpredictable and unrelated to dose, and the bone marrow does not usually recover when the agent is withdrawn.4 Documentation of an identifiable factor or agent inducing aplastic anemia in these cases is difficult, because evidence is primarily circumstantial and symptoms may occur months or years after exposure. Some drugs associated with idiosyncratic, acquired aplastic anemia are listed in Box 21-3.4,8 Aplastic anemia as an idiosyncratic adverse reaction to a drug, chemical, or other agent is a rare event and is likely due to a combination of genetic and environmental factors in susceptible individuals. Currently no tests are available to predict individual susceptibility to these idiosyncratic reactions; however, genetic variations in immune response pathways or metabolic enzymes may play a role.4 There is an approximately twofold higher incidence of HLA-DR2 and its major serologic split, HLA-DR15, in aplastic anemia patients than in the general population, but the relationship of this finding to the disease pathophysiology has not been elucidated.9,10 Genetic polymorphisms in enzymes that metabolize benzene may increase an individual’s susceptibility to its toxicity, even at low levels of exposure.4,11 These include polymorphisms in glutathione S-transferase (GST) enzymes (GSTT1 and GSTM1), myeloperoxidase, nicotinamide adenine dinucleotide phosphate (reduced form):quinine oxidoreductase 1, and cytochrome oxidase P450 2E1.11 A deficiency in GST due to the GSTT1 null genotype is overrepresented in white, Hispanics, and Asians with aplastic anemia with a frequency of 30%, 28%, and 75%, respectively.12 White patients with aplastic anemia also have a higher frequency (22%) of the GSTM1/GSTT1 null genotype than the general population.12 GST is important for metabolism of chemical toxins, and deficiencies of this enzyme may increase the risk of developing aplastic anemia. Further study is required to assess how these genetic variations, and other factors yet to be discovered, contribute to the bone marrow suppression in aplastic anemia. Acquired aplastic anemia occurs occasionally as a complication of infection with Epstein-Barr virus, human immunodeficiency virus (HIV), hepatitis virus, and human parvovirus B19.4 A history of acute non-A, non-B, or non-C hepatitis 1 to 3 months before onset of pancytopenia is found in 2% to 10% of patients with acquired aplastic anemia.13 Posthepatitis aplastic anemia syndrome has a poor prognosis. Aplastic anemia associated with pregnancy is a rare occurrence, with fewer than 100 cases reported in the literature.14 Approximately 10% of individuals with acquired aplastic anemia have a concomitant autoimmune disease,15 and approximately 10% develop hemolytic or thrombotic manifestations of paroxysmal nocturnal hemoglobinuria (PNH).16 The overlap between acquired aplastic anemia and PNH is discussed later. The primary lesion in acquired aplastic anemia is a quantitative and qualitative deficiency of hematopoietic stem cells. In culture, stem cells of patients with acquired aplastic anemia have diminished colony formation.17 The hematopoietic stem and early progenitor cell compartment is identified by expression of CD34 surface antigens. When measured by flow cytometry, the CD34+ cell population in the bone marrow of patients with aplastic anemia can be 10 times lower than that in healthy individuals.17 In addition, these CD34+ cells have an increased expression of Fas receptors that mediate apoptosis18,19 and an increased expression of apoptosis-related genes as determined by microarray analysis.20 The bone marrow stromal cells are functionally normal in acquired aplastic anemia. They produce normal or increased quantities of growth factors, and they are able to support the growth of CD34+ cells from healthy donors in culture and in vivo after transplantation.4,21 Individuals with aplastic anemia have elevated serum levels of erythropoietin, thrombopoietin, granulocyte colony-stimulating factor (G-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF).22 In addition, the serum level of FLT3 ligand, a growth factor that stimulates proliferation of stem and progenitor cells, is up to 200 times higher in patients with severe aplastic anemia than in healthy controls.22,23 Despite their elevated levels, growth factors alone are generally unsuccessful in correcting the cytopenias found in acquired aplastic anemia. The severe depletion of hematopoietic stem and progenitor cells from the bone marrow may be due to (1) direct damage to stem cells, (2) immune damage to stem cells, or (3) other unknown mechanisms. In the direct mechanism, a cytotoxic drug, chemical, radiation, or virus damages the DNA of the stem and progenitor cells, causing apoptosis and cytolysis.4 In the immune mechanism, exposure to certain drugs, chemicals, viruses, or other unknown agents causes an autoimmune cytotoxic T lymphocyte attack that destroys the stem and progenitor cells.24 An autoimmune pathophysiology was first suggested in the 1970s when aplastic anemia patients undergoing immunosuppressive conditioning before bone marrow transplantation experienced an improvement in cell counts.25 Evidence supporting an autoimmune pathophysiology is that in immune-related acquired aplastic anemia (1) there is an increase in cytotoxic (CD8+) T lymphocytes in the blood and bone marrow with an oligoclonal expansion of specific T-cell clones26; (2) these T cells produce increased amounts of cytokines, such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), that inhibit hematopoiesis and induce apoptosis27–29; they also have an upregulation of T-bet, a transcription factor that binds to the promoter of the IFN-γ gene30; (3) there is an increase in TNF-α receptors on CD34+ cells31; and (4) there is improvement in the cytopenia after immunosuppressive therapy (IST).4,24 Approximately two thirds of patients with acquired aplastic anemia respond to IST; the one third who do not respond may have a severely depleted stem cell compartment or other immune or nonimmune pathophysiologic factors may be present.32 Possible mechanisms by which a drug, chemical, or virus may elicit an autoimmune response include mutation or alteration of stem cell antigens and disruption of immune regulation mechanisms. Concerning the former mechanism, a drug, chemical, or virus may alter self-proteins, induce expression of abnormal or novel antigens, expose hidden antigens, or induce an immune response that cross-reacts with self-antigens.24,28 With regard to the latter mechanism, CD4+CD25+FOXP3+ regulatory T cells are decreased in aplastic anemia.33 Regulatory T cells normally suppress autoreactive T cells, and a deficit of these cells may facilitate an autoimmune reaction. In addition, 36.9%, 10.9%, 21.9%, and 43.8% of individuals with aplastic anemia have the single nucleotide polymorphisms IFN-γ/+874 TT, TNF-α/–308 AA, transforming growth factor-β1/–509 TT, and interleukin-6/–174 GG, respectively.34 These polymorphisms result in cytokine overproduction and may impart a genetic susceptibility to aplastic anemia and contribute to its severity.34 The antigens responsible for triggering and sustaining the autoimmune attack on the stem cells are unknown. Possible autoantigens have been identified using sera from aplastic anemia patients; these include kinectin,35 diazepam-binding inhibitor-related protein 1,36 and moesin.37 These proteins are expressed in hematopoietic progenitor cells, but their role in the pathogenesis of aplastic anemia requires further investigation. Approximately one third of patients with acquired aplastic anemia have shortened telomeres in their peripheral blood granulocytes compared with aged-matched controls.38,39 Telomeres protect the ends of chromosomes from damage and erosion, and cells with abnormally short telomeres undergo proliferation arrest and premature apoptosis. Approximately 10% of patients with acquired aplastic anemia and shortened telomeres have a mutation in the gene for either the ribonucleic acid (RNA) template (TERC) or the reverse transcriptase (TERT) of the telomerase complex.39–41 Telomerase is an enzyme complex that repairs and maintains the telomeres. The reason for the shortened telomeres in the other 90% of patients is unknown, but may be the stressed hematopoiesis brought on by the autoimmune attack or other unknown mutations.39 In stressed hematopoiesis, there is an increase in progenitor cell turnover, and the telomeres become shorter with each cell division. About 4% of patients with acquired aplastic anemia and short telomeres have mutations in the SBDS gene.42 The SBDS gene product is involved in ribosome biogenesis, but its relationship to telomere maintenance is unknown.42 TERT/TERC and SBDS mutations occur in the inherited aplastic anemias dyskeratosis congenita and Shwachman-Diamond syndrome, respectively, and some acquired aplastic anemia patients with these mutations may actually have dyskeratosis congenita or Shwachman-Diamond syndrome with an atypical presentation.3,4 Identification of these genetic defects in patients with acquired aplastic anemia has important implications for treatment in that IST may not be effective and apparently healthy HLA-matched siblings who test positive for the same genetic mutation should not be selected as donors for bone marrow transplantation.39 Shortened telomeres occur more often in patients whose pancytopenia does not respond to IST,43 and a defect in telomere maintenance may be another pathophysiologic mechanism of stem cell depletion, imparting a susceptibility to bone marrow failure after an environmental insult with a virus, drug, or autoimmune attack.39,41 Pancytopenia is typical, although initially only one or two cell lines may be decreased. The absolute neutrophil count is decreased, and the absolute lymphocyte count may be normal or decreased. The hemoglobin is usually less than 10 g/dL, the mean cell volume (MCV) is increased or normal, and the percent and absolute reticulocyte counts are decreased. Table 21-1 lists the diagnostic criteria for aplastic anemia by degree of severity.6,8,44,45 This classification is helpful in guiding treatment decisions. TABLE 21-1 Diagnostic Criteria for Aplastic Anemia *Or 25% to 50% cellularity with <30% residual hematopoietic cells. In the peripheral blood, neutrophils, monocytes, and platelets are decreased, and the RBCs are macrocytic or normocytic (Figure 21-1). Toxic granulation may be observed in the neutrophils, but the RBCs and platelets are normal in appearance. Blasts and other immature blood cells are characteristically absent. Serum iron level and percent transferrin saturation are increased, which reflects the decreased use of iron for erythropoiesis. Liver function test results may be abnormal if the pancytopenia was preceded by hepatitis. When high-resolution flow cytometry is used, about two thirds of patients are found to have small numbers of PNH granulocytes (less than 25%) in the peripheral blood,46 but only about 10% of patients develop a large enough clone of PNH cells to display the typical clinical and biochemical manifestations of PNH.16 PNH is characterized by an acquired stem cell mutation that results in circulating blood cells that lack glycosylphosphatidylinositol (GPI)–linked proteins, such as CD55 and CD59. The absence of CD55 and CD59 on the surface of the RBCs renders them more susceptible to lysis by complement. The significance of the circulating PNH cells in aplastic anemia is uncertain, but it may be related to the ability of the GPI-deficient stem cells to escape immune destruction.24,32 It is important to test for PNH cells in acquired aplastic anemia because of the hemolytic and/or thrombotic complications associated with PNH (see Chapter 23). The Ham test for complement-mediated hemolysis is positive only if there are large numbers of circulating PNH cells; however, flow cytometry is a more sensitive method and has replaced the Ham test for diagnosis of PNH (see Chapter 23).8,46,47 Bone marrow aspirates and biopsy specimens have prominent fat cells with areas of patchy cellularity. Biopsy samples are required for an accurate quantitative assessment of the marrow cellularity, and severe hypocellularity is a characteristic feature (Figure 21-2). Erythroid, granulocytic, and megakaryocytic cells are decreased or absent. Dyserythropoiesis may be present, but there is no dysplasia of the granulocyte or platelet cell lines. Blasts and abnormal cell infiltrates are characteristically absent. Reticulin staining is normal. In patients receiving IST, the risk of developing an abnormal karyotype is 14% at 5 years and 20% at 10 years.48 Monosomy 7 and trisomy 8 are the most frequently identified cytogenetic abnormalities.47,48 Cytogenetic analysis using conventional culture techniques underestimates the incidence of karyotype abnormalities because of the hypocellularity of the bone marrow and scarcity of cells in metaphase.49 Interphase fluorescent in situ hybridization (FISH) using deoxyribonucleic acid (DNA) probes for specific chromosome abnormalities has the advantage of greater sensitivity and the ability to use nondividing cells.49 In one study, FISH detected monosomy 7 or trisomy 8 in 26% of aplastic anemia patients who had a normal karyotype by conventional cytogenetic testing.49 Patients with inherited aplastic anemia may be diagnosed erroneously as having acquired aplastic anemia if the disease first becomes apparent in late adolescence or adulthood or if they lack the typical clinical and physical manifestations.3,4 Exclusion of inherited aplastic anemias in the differential diagnosis of acquired aplastic anemia is essential, because these conditions require a different therapeutical approach. The inherited aplastic anemias are discussed later in the chapter. Severe aplastic anemia requires immediate treatment to prevent the dire consequences of serious pancytopenia. If a potential causative agent is suspected, its use should be discontinued. To keep blood counts at safe levels, platelets generally are administered when the platelet count decreases to less than 10 × 109/L, whereas RBCs are given when the patient becomes symptomatic.8 One of the most important early decisions that must be made is whether a patient is a candidate for bone marrow transplantation. Bone marrow transplantation is generally the treatment of choice for patients with severe aplastic anemia who are younger than age 40 and have an HLA-identical sibling.4,8 Unfortunately, only 20% to 30% of patients meet these criteria.4 For patients older than age 40 and those of any age without an HLA-identical sibling, IST, consisting of antithymocyte globulin with cyclosporine, is the preferred therapy.8,16 The antithymocyte globulin decreases the number of activated T cells, and the cyclosporine inhibits their function, suppressing the autoimmune reaction against the stem cells. Approximately two thirds of patients initially respond to IST but 30% to 40% eventually experience relapse.16,32
Bone Marrow Failure
Case Study
Patient
Reference Range
WBCs (× 109/L)
2.0
4.5-11.0
Hb (g/dL)
8.0
12.0-15.0
MCV (fL)
104
80-100
Platelets (× 109/L)
27
150-450
Reticulocytes (%)
0.6%
0.5-1.5
Reticulocytes (× 109/L)
16
25-75
Neutrophils (× 109/L)
1.1
2.3-8.1
Lymphocytes (× 109/L)
0.4
0.8-4.8
Aplastic Anemia
Acquired Aplastic Anemia
Incidence
Etiology
Pathophysiology
Laboratory Findings
NSAA
SAA
VSAA
Bone marrow
Hypocellular bone marrow plus at least two of the following:
Bone marrow cellularity <25%* plus at least two of the following:
Same as SAA
Neutrophils (× 109/L)
0.5-1.5
0.2-0.5
<0.2
Platelets (× 109/L)
20-50
<20
Same as SAA
Other
Hb ≤10 g/dL plus reticulocytes <30 × 109/L
Reticulocytes <20 × 109/L or <1% corrected for Hct
Same as SAA

Treatment and Prognosis
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Bone Marrow Failure