Chapter 21 Blood cell antigens and antibodies
erythrocytes, platelets and granulocytes
Erythrocytes
Red Cell Antigens
A total of 30 blood group systems have been described (Table 21.1). Each system is a series of red cell antigens, determined either by a single genetic locus or very closely linked loci. In addition to the blood group systems, there are six ‘collections’ of antigens (e.g. Cost), which bring together other genetically, biochemically or serologically related sets of antigens and a separate series of low-frequency (e.g. Rd) and high-frequency (e.g. Vel) antigens, which do not fit into any system or collection. A numeric catalogue of red cell antigens is being maintained by an International Society of Blood Transfusion (ISBT) Working Party.1
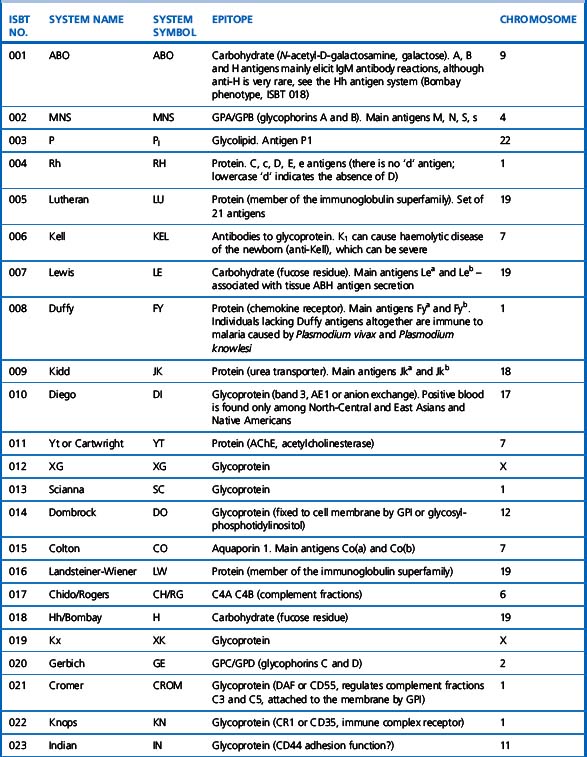
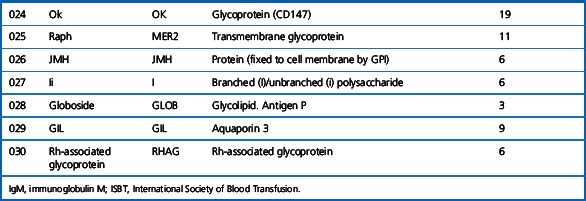
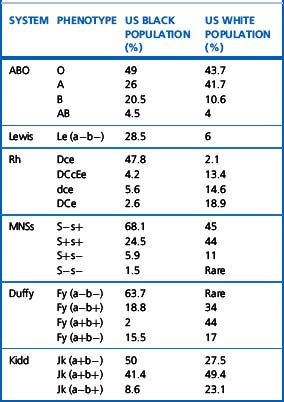
Alternative forms of a gene coding for red cell antigens at a particular locus are called alleles and individuals may inherit identical or non-identical alleles. Most blood group genes have been assigned to specific chromosomes (e.g. ABO system on chromosome 9, Rh system on chromosome 1). The term genotype is used for the sum of the inherited alleles of a particular gene (e.g. AA, AO) and most red cell genes are expressed as codominant antigens (i.e. both genes are expressed in the heterozygote). The phenotype refers to the recognizable product of the alleles and there are many racial differences in the frequencies of red cell phenotypes, as shown in Table 21.2.
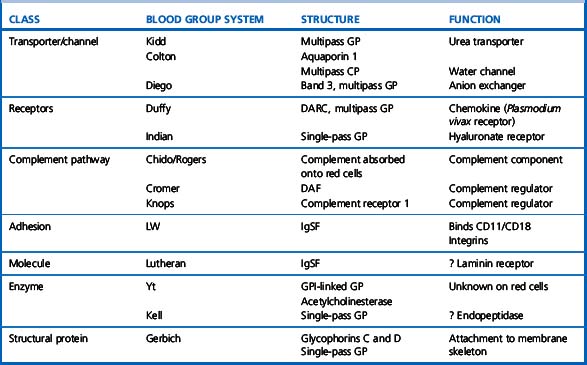
Red cell antigens are determined either by carbohydrate structures or protein structures. Carbohydrate-defined antigens are indirect gene products (e.g. ABO, Lewis, P). The genes code for an intermediate product, usually an enzyme that creates the antigenic specificity by transferring sugar molecules onto the protein or lipid. Protein-defined antigens are direct gene products and the specificity is determined by the inherited amino acid sequence and/or the conformation of the protein. Proteins carrying red cell antigens are inserted into the membrane in one of three ways: single pass, multipass or linked to phosphatidylinositol (GPI-linked). Only a few red cell antigens are erythroid-specific (Rh, LW, Kell and MNSs), the remainder being expressed in many other tissues. The structure and functions of the membrane proteins and glycoproteins carrying blood group antigens have been reviewed by Daniels.2 An illustration of the putative functions of molecules containing blood group antigens is provided in Table 21.3.
ABO System
Discovery of the ABO system by Landsteiner marked the beginning of safe blood transfusion. The ABO antigens, although most important in relation to transfusion, are also expressed on most endothelial and epithelial membranes and are important histocompatibility antigens.3 Transplantation of ABO-incompatible solid organs increases the potential for hyperacute graft rejection, although ABO-incompatible renal transplantation can be successfully carried out with plasmapheresis in addition to immunosuppression of the recipient.4 Major ABO-incompatible stem cell transplants (e.g. group A stem cells into a group O recipient) will provoke haemolysis, unless the donation is depleted of red cells.
ABO Antigens and Encoding Genes
There are four main blood groups: A, B, AB and O (Table 21.4). In the British Caucasian population, the frequency of group A is 42%, B 9%, AB 3% and O 46%, but there is racial variation in these frequencies.5 The epitopes of ABO antigens are determined by carbohydrates (sugars), which are linked either to polypeptides (forming glycoproteins) or to lipids (glycolipids).
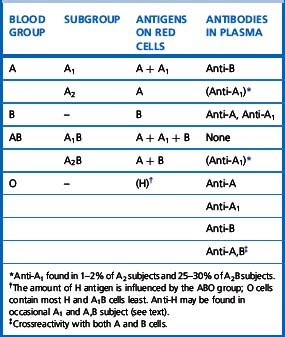
The expression of ABO antigens is controlled by three separate genetic loci: ABO located on chromosome 9 and FUT1 (H) and FUT2 (Se), both of which are located on chromosome 19. The genes from each locus are inherited in pairs as Mendelian dominants. Each gene codes for a different enzyme (glycosyltransferase), which attaches specific monosaccharides onto precursor disaccharide chains (Table 21.5). There are four types of disaccharide chains known to occur on red cells, on other tissues and in secretions. The Type 1 disaccharide chain is found in plasma and secretions and is the substrate for the FUT2 (Se) gene, whereas Types 2, 3 and 4 chains are only found on red cells and are the substrate for the FUT1 (H) gene. It is likely that the O and B genes arose by mutation of the A gene. The O gene does not encode for the production of a functional enzyme; group O individuals commonly have a deletion at nucleotide 261 (the O1 allele), which results in a frame-shift and premature termination of the translated polypeptide and the production of an enzyme with no catalytic activity. The B gene differs from A by consistent nucleotide substitutions.6 The expression of A and B antigens is dependent on the H and Se genes, which both give rise to glycosyltransferases that add L-fucose, producing the H antigen. The presence of an A or B gene (or both) results in the production of further glycosyltransferases, which convert H substance into A and B antigens by the terminal addition of N-acetyl-D-galactosamine and D-galactose, respectively (Fig. 21.1). Because the O gene produces an inactive transferase, H substance persists unchanged as group O. In the extremely rare Oh Bombay phenotype, the individual is homozygous for the h allele of FUT1 and hence cannot form the H precursor of the A and B antigen. Their red cells type as group O, but their plasma contains anti-H, in addition to anti-A, anti-B and anti-A,B, which are all active at 37°C. As a consequence, individuals with an Oh Bombay phenotype can only be safely transfused with other Oh red cells.
Table 21.5 Glycosyltransferases produced by genes encoding antigens within the ABO, H and Lewis blood group systems
| Gene | Allele | Transferase |
|---|---|---|
| FUT1 | H | α-2-L-fucosyltransferase |
| h | None | |
| A | A | α-3-N-acetyl-D-galactosaminyltransferase |
| B | B | α-3-D-galactosyltransferase |
| O | O | None |
| FUT2 | Se | α-2-L-fucosyltransferase |
| se | None | |
| FUT3 | Le | α-3/4-L-fucosyltransferase |
| le | None |
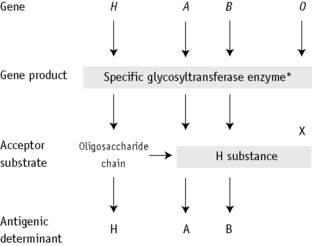
Serologists have defined two common subgroups of the A antigen. Approximately 20% of group A and group AB individuals belong to group A2 and group A2B, respectively, the remainder belonging to group A1 and group A1B. These subgroups arise as a result of inheritance of either the A1 or A2 alleles. The A2 transferase is less efficient in transferring N-acetyl-D-galactosamine to available H antigen sites and cannot utilize Types 3 and 4 disaccharide chains. As a consequence, A2 red cells have fewer A antigen sites than A1 cells and the plasma of group A2 and group A2B individuals may also contain anti-A1. The distinction between these subgroups can be made using the lectin Dolichos biflorus, which only reacts with A1 cells. The H antigen content of red cells depends on the ABO group and, when assessed by agglutination reactions with anti-H, the strength of reaction tends to be graded O > A2 > A2B > B > A1 > A1B. Other subgroups of A are occasionally found (e.g. A3, Ax) that result from mutant forms of the glycosyltransferases produced by the A gene and are less efficient at transferring N-acetyl-D-galactosamine onto H substance.6
Secretors and Non-Secretors
The ability to secrete A, B and H substances in water-soluble form is controlled by FUT2 (dominant allele Se). In a Caucasian population, about 80% are secretors (genotype SeSe or Sese) and 20% are non-secretors (genotype sese) (Table 21.6). Secretors have H substance in the saliva and other body fluids together with A substances, B substances or both, depending on their blood group. Only traces of these substances are present in the secretions of non-secretors, although the antigens are expressed normally on their red cells and other tissues.
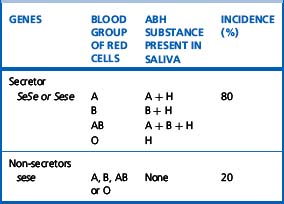
An individual’s secretor status can be determined by testing for ABH substance in saliva (see p. 504).
ABO Antigens and Disease
Group A individuals rarely may acquire a B antigen from a bacterial infection that results in the release of a deacetylase enzyme. This converts N-acetyl-D-galactosamine into α-galactosamine, which is similar to galactose, the immunodominant sugar of group B, thereby sometimes causing the red cells to appear to be group AB. In the original reported cases, five out of seven of the patients had carcinoma of the gastrointestinal tract. Case reports attest to the danger of individuals with an acquired B antigen being transfused with AB red cells, resulting in a fatal haemolytic transfusion reaction following the production of hyperimmune anti-B.7
The inheritance of ABH antigens is also known to be weakly associated with predisposition to certain diseases. Group A individuals have 1.2 times the risk of developing carcinoma of the stomach than group O or B; group O individuals have 1.4 times more risk of developing peptic ulcer than non-group O individuals; and non-secretors of ABH have 1.5 times the risk of developing peptic ulcer than secretors.8 The ABO group also affects plasma von Willebrand factor (VWF) and factor VIII levels; group O healthy individuals have levels around 25% lower than those of other ABO groups.9 ABO blood group appears to mediate its effect by accelerating clearance of VWF but the mechanism is not yet clear10 ABH antigens are also frequently more weakly expressed on the red cells of persons with leukaemia.
ABO Antibodies
Anti-A and anti-B
ABO antibodies, in the absence of the corresponding antigens, appear during the first few months after birth, probably as a result of exposure to ABH antigen-like substances in the diet or the environment (i.e. they are ‘naturally occurring’) (Table 21.4). This allows for reverse (serum/plasma) grouping as a means of confirming the red cell phenotype. The antibodies are a potential cause of dangerous haemolytic transfusion reactions if transfusions are given without regard to ABO compatibility. Anti-A and anti-B are always, to some extent, immunoglobulin M (IgM). Although they react best at low temperatures, they are nevertheless potentially lytic at 37°C. Hyperimmune anti-A and anti-B occur less frequently, usually in response to transfusion or pregnancy, but they may also be formed following the injection of some toxoids and vaccines. They are predominantly of IgG class and are usually produced by group O and sometimes by group A2 individuals. Hyperimmune IgG anti-A and/or anti-B from group O or group A2 mothers may cross the placenta and cause haemolytic disease of the newborn (HDN). These antibodies react over a wide thermal range and are more effective haemolysins than the naturally occurring antibodies. Group O donors should always be screened for high-titre anti-A and anti-B antibodies, which may cause haemolysis when group O platelets or plasma are transfused to recipients with A and B phenotypes.
Anti-A1 and anti-H
Anti-H reacts most strongly with group O and A2 red cells and also normally acts as a cold agglutinin. A notable, but rare, exception is the anti-H that occurs in the Oh Bombay phenotype, which is an IgM antibody and causes lysis at 37°C (Table 21.4) so that Oh Bombay phenotype blood would be required for transfusion.
Rh System
Rh Antigens and Encoding Genes
In Caucasian, D-negative individuals, the RHD gene is deleted, whereas in Black races and other populations, single-point mutations, partial deletions or recombinations have been described. In individuals with a weak D antigen (Du), there is a quantitative reduction in D antigen sites, believed to arise from an uncharacterized transcriptional defect. These individuals do not make anti-D antibodies following a D antigen challenge. Partial D individuals lack one or more epitopes of the D antigen, defined using panels of monoclonal reagents. Dvi is perhaps the most important partial D phenotype because such individuals not infrequently make anti-D. Partial D phenotypes arise from DNA exchanges between RHD and RHCE genes and from other rearrangements. Comprehensive reviews of this system have been provided by Avent and Reid11 and Daniels et al.12
The Rh haplotypes are named either by the component antigens (e.g. CDe, cde) or by a single shorthand symbol (e.g. R1 = CDe, r = cde). Thus, a person may inherit CDe (R1) from one parent and cde (r) from the other and have the genotype CDe/cde (R1r). The haplotypes in order of frequency and the corresponding shorthand notation are given in Table 21.7. Although two other nomenclatures are also used to describe the Rh system, namely, Wiener’s Rh-Hr terminology and Rosenfield’s numeric notation, the CDE nomenclature, derived from Fisher’s original theory, is recommended by a World Health Organization Expert Committee13 in the interest of simplicity and uniformity. The Rh antigens are defined by corresponding antisera, with the exception of ‘anti-d’, which does not exist. Consequently, the distinction between homozygous DD and the heterozygous Dd cannot be made by direct serological testing but may be resolved by informative family studies. It is still routine practice to predict the genotype from the phenotype on the basis of probability tables for the various Rh genotypes in the population (Table 21.7). However, in women with immune anti-D and a history of an infant affected by HDN, RH DNA typing is used in prenatal testing for the fetal D status to decide on the clinical management of the pregnancy, e.g. the need for monitoring for fetal anaemia using middle cerebral artery Doppler ultrasound. Suitable sources include amniotic fluid (amniocytes) and trophoblastic cells (chorionic villi) or after 15 weeks’ gestation, maternal blood can be used because it contains fetal DNA.14,15 In practice, multiplex polymerase chain reaction (PCR) is used, with more than two primer sets, to detect the different molecular bases for D-negative phenotypes in non-Caucasians. RH DNA typing also has applications in paternity testing and forensic medicine. There are racial differences in the distribution of Rh antigens, e.g. D negativity is more common in Caucasians (approximately 15%), whereas Ro (cDe) is found in approximately 48% of Black Americans but is uncommon (approximately 2%) in Caucasians. The Rh antigens are present only on red cells and are a structural part of the cell membrane. Complete absence of Rh antigens (Rh-null phenotype) may be associated with a congenital haemolytic anaemia with spherocytes and stomatocytes in the blood film, increased osmotic fragility and increased cation transport. This phenotype arises either as a result of homozygosity for silent alleles at the RH locus (the amorph type) or more commonly by homozygosity for an autosomal suppressor gene (X), genetically independent of the RH locus (the regulator type). Rh antigens are well-developed before birth and can be demonstrated on the red cells of very early fetuses.
Table 21.7 The Rh haplotypes in order of frequency (Fisher nomenclature) in Caucasians and the corresponding short notations
| Fisher | Short Notations | Approximate Frequency (%) |
|---|---|---|
| CDe | R1 | 41 |
| cde | r | 39 |
| cDE | R2 | 14 |
| cDe | R0 | 3 |
| CwDe | R1w | 1 |
| cdE | r″ | 1 |
| Cde | r′ | 1 |
| CDE | RZ | Rare |
| CdE | ry | Rare |
MNSs System
Clinical Significance of Red Cell Alloantibodies
The significance of the alloantibodies described, with respect to the nature of the haemolytic transfusion reaction they produce, is provided in Table 21.8. The majority of haemolytic transfusion reactions, however, are the result of ABO incompatibility.16
Table 21.8 Antibody specificities related to the mechanism of immune haemolytic destruction
| Blood Group System | Intravascular Haemolysis | Extravascular Haemolysis |
|---|---|---|
| ABO,H | A, B, H | |
| Rh | All | |
| Kell | K | K, k, Kpa, Kpb, Jsa, Jsb |
| Kidd | Jka | Jka, Jkb, Jk3 |
| Duffy | Fya, Fyb | |
| MNS | M, S, s, U | |
| Lutheran | Lub | |
| Lewis | Lea | |
| Cartwright | Yta | |
| Colton | Coa, Cob | |
| Dombrock | Doa, Dob |
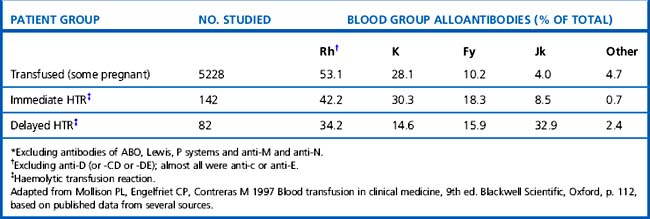
Mollison et al.17 analysed the significance of blood group antigens other than those of the ABO system and D by looking at the prevalence of transfusion-induced red cell alloantibodies, excluding anti-D, -CD and -DE (Table 21.9). Rh antibodies, mainly anti-c or anti-E, accounted for 53% of the total and anti-K and anti-Fya accounted for a further 38%, leaving only about 9% for all other specificities. A similar distribution of the different red cell antibodies was found in a smaller group of patients who experienced immediate haemolytic transfusion reactions (HTR). However, the figures for delayed HTR showed a striking increase in the relative frequency of Jk antibodies, which reflects the outlined characteristics of Jk antibodies.
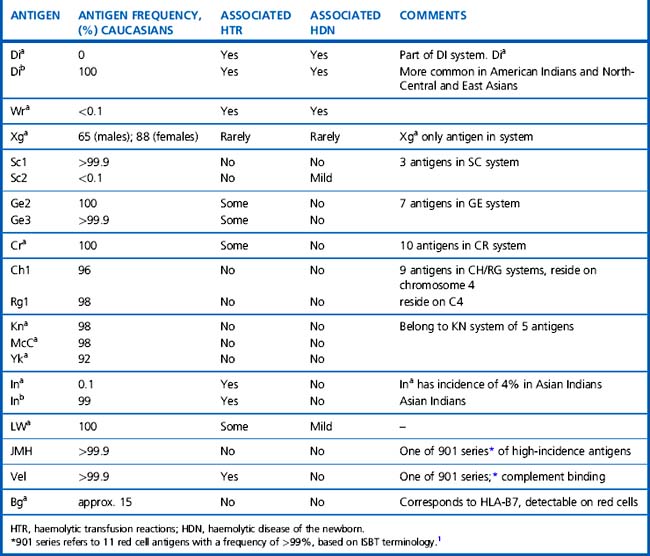
The significance of the many other blood group antigens not referred to in the text is summarized in Table 21.10. However, it should be noted that the antibodies listed are usually wholly or predominantly IgG and would be detectable in routine pretransfusion testing using the indirect anti-globulin test (IAT).
Mechanisms of Immune Destruction of Red Cells
Immune-mediated haemolysis of red cells depends on the following:18
The mechanism of immune haemolysis also determines the site of haemolysis:
Macrophage activity is an important component of cell destruction and further study of cellular interactions at this stage of immune haemolysis may provide an explanation for the differing severity of haemolysis in patients with apparently similar antibodies. In vitro macrophage (monocyte) assays have been used sometimes to supplement conventional serological techniques to assess this aspect of immune haemolysis.20
Interleukin-6 also enhances FcγRII activation and increased activity of the CR1 receptor occurs through the action of T-cell cytokines and through chemotactic agents released in the inflammatory response.22 The increased levels of proinflammatory cytokines and other biological mediators and their effects on the activity of the monocyte phagocytic system have been monitored in patients with systemic inflammatory response syndrome.23 It is therefore possible that release of cytokines during viral and bacterial infections could, at least in part, trigger some episodes of autoimmune cell destruction.
Antigen–Antibody Reactions
Some complement-binding antibodies (especially IgM) may cause lysis in vitro (without noticeable agglutination), which can be enhanced by the addition of fresh serum as a source of complement. However, complement activation may only proceed to the C3 stage; in these circumstances cell-bound C3 can be detected by the antiglobulin test using an appropriate anticomplement reagent (see p. 500).
Quality Assurance within the Laboratory
It has long been appreciated that the test systems used for routine pre-transfusion testing are of the utmost importance because errors can and do lead to patient morbidity and mortality. It is therefore of little surprise that within the European Union all reagents, calibrators and control materials for red cell typing and for determining the presence of ‘irregular anti-erythrocytic antibodies’ have been included under the In-vitro Diagnostics (IVD) Medical Devices Directive24 (see p. 588). This means that all reagents sold within the European Union must display the CE mark to show that they conform to the agreed Common Technical Specifications (CTS). In each European country, a Competent Authority will be able to withdraw or suspend certification of any reagent, depending on the information received from its Notified Body, which will perform batch release approval and monitor the performance of the manufacturer and the product.
Related posts:
 Preparation and staining methods for blood and bone marrow films
Investigation of a thrombotic tendency
Laboratory methods used in the investigation of the haemolytic anaemias
Laboratory organization and management
Investigation of the hereditary haemolytic anaemias: membrane and enzyme abnormalities
Diagnostic radioisotopes in haematology
Preparation and staining methods for blood and bone marrow films
Investigation of a thrombotic tendency
Laboratory methods used in the investigation of the haemolytic anaemias
Laboratory organization and management
Investigation of the hereditary haemolytic anaemias: membrane and enzyme abnormalities
Diagnostic radioisotopes in haematology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


