Biomarker-based clinical trial design in the era of genomic medicine
Richard Simon, DSc  Martine Piccart-Gebhart, MD, PhD
Martine Piccart-Gebhart, MD, PhD
Overview
The increasingly recognized molecular heterogeneity of human cancers together with the growing availability of powerful tools providing “molecular portraits” of this heterogeneity require the development of new paradigms for the development of a reliable basis for “personalized” medicine. We review new approaches to the design and analysis of clinical trials for the development of “targeted” therapeutics and diagnostics to inform their use.
Introduction: why we need new designs and analysis paradigms for biomarker-driven clinical trials
Randomized clinical trials have enabled the development of a reliable evidence-based medicine. They have generally evaluated a treatment relative to a control regimen for a broadly defined population of patients defined by primary site, histology, stage, and number of prior treatments. In recent years, sequencing of human tumor DNA has established that cancers of a primary site often represent a heterogeneous collection of diseases that differ with regard to the genomic aberrations that cause them and drive their invasion.1 The tumors are often heterogeneous with regard to the genes and pathways that can be inhibited by molecularly targeted therapy. Broad eligibility clinical trials are severely underpowered for detecting benefit for the subset of patients whose tumors are most susceptible to the action of the new drug. In the broad eligibility clinical trials that are “positive” because the sample size has been increased to such an extent that even small average treatment effects are statistically significant, only a small proportion of the intent to treat population actually benefits from treatment resulting in toxicity for patients who do not benefit and a large societal economic cost for overtreatment of the population.
Today, we have powerful tools for characterizing the tumors biologically and using this characterization to prospectively structure the design and analysis of more informative clinical trials that result in larger treatment effects for a larger portion of the treated patients. In this chapter, we discuss these modern designs and analyses that should better respond to the needs of “personalized” oncology.
Phase II trials of molecularly targeted agents with companion diagnostics
Nowadays, most of the cancer drugs are being developed for defined molecular targets. In some cases, the targets are well understood, and there is a compelling biological basis and evidence from phase I clinical trials for restricting development to the subset of patients whose tumors are characterized by deregulation of the drug target. For other drugs, there are multiple targets and more uncertainty about how to measure whether a drug target is driving tumor biology in an individual patient.2
The ideal approach in the former situation is the codevelopment of the drug and a companion diagnostic that measures the deregulation of the drug target in a robust way.
In the era of molecular oncology, not only must the phase II trial determine whether there is activity of the drug overall in a histologic type, but it also must determine whether the subsequent phase III trial should be restricted based on a candidate companion diagnostic. Freidlin et al.3 have described a design for use with a single binary candidate biomarker in a randomized phase II design. Their design enables one to determine whether the drug should be (1) developed in a phase III enrichment trial, (2) developed in an analysis stratified all comers’ trial, (3) developed in an all comers’ trial without measurement of the biomarker, or (4) dropped from further development. This design is shown in Figure 1. The sample size is determined so that the treatment effect on progression-free survival (PFS) in the marker-positive subset has power 0.9 for detecting a doubling of median PFS at a one-sided significance level of 0.10. This will generally provide at least as many events for evaluating treatment effect in the marker-negative subset. If the treatment effect in the marker-positive subset is not significant, then the overall treatment effect is tested at a one-sided 0.05 significance level. If that treatment effect is not statistically significant, then the drug is not recommended for phase III development. If the overall treatment effect is significant, then a traditional phase III trial without measurement of the candidate biomarker is recommended. If the treatment effect in the marker-positive subset is significant, then one examines an 80% confidence interval for the hazard ratio of treatment effect in the marker-negative subset. If the upper confidence interval is below 1.3, then one concludes that the treatment is not active in the marker-negative patients and an enrichment phase III trial is recommended. If the lower confidence limit is above 1.5, then one concludes that the treatment works well in the marker-negative patients and a traditional phase III trial in which the biomarker is not even measured is recommended. Otherwise, a biomarker-stratified phase III trial is recommended.
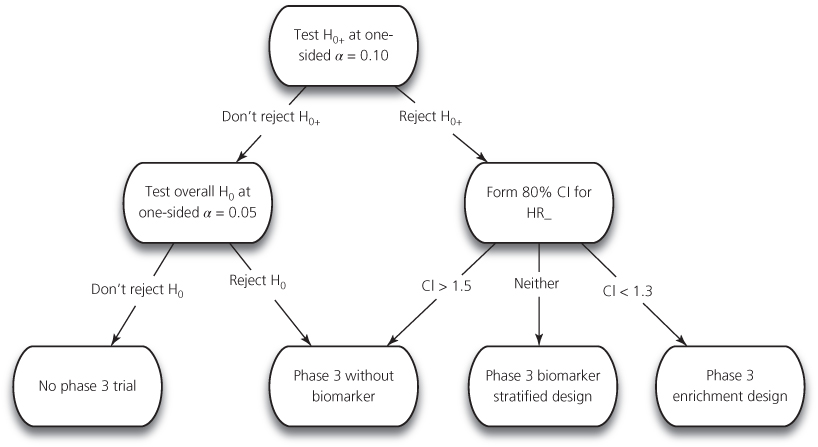
Figure 1 Analysis strategy for randomized phase II design to decide whether phase III trial should be restricted to biomarker-positive population, should include positives and negatives, whether biomarker should not even be measured or whether the conduct of a phase III trial is not supported by phase II results. Data from [3]. HR+ is hazard ratio in biomarker-positive group. HR− is hazard ratio for treatment in biomarker-negative group. H0+ is null hypothesis that HR+ = 1. H0 is null hypothesis that overall HR = 1.
Other phase II designs for evaluation of a treatment within marker subsets in single-arm phase II studies have been described by Pusztai and Hess4 and Jones and Holmgren.5 These designs are focused primarily on ensuring that promising activity of the drug is not missed in cases where its activity is restricted to test-positive patients, and yet excessive numbers of patients are not required in cases where its activity is sufficiently broad that the marker is not needed.
There are more complicated phase II settings, where no natural cut-point of the biomarker is known in advance, or where there are multiple candidate biomarkers. The BATTLE I trial in NSCLC (nonsmall cell lung cancer) is an example of a phase II clinical trial using a response-adaptive randomization,6 which is discussed in another chapter of this book. Of note, some statisticians have raised doubts about the effectiveness of such adaptive randomization designs.7
Phase IIa basket and umbrella discovery trials
Large tumor sequencing studies such as the Cancer Genome Project in the United Kingdom and the Cancer Genome Atlas (TCGA) in the United States have identified recurrent genomic aberrations in a variety of primary tumor sites.1 These data provide a scientific basis for treatment of individual patients based on the biological characterization of their tumors. There are, however, many challenges in moving tumor genomics to clinical oncology and the first one is the tremendous heterogeneity of the genomic landscapes of the tumors, implying the existence of multiple, rare genomic segments within a given tumor type.
Developing a single targeted drug in a single small genomic segment of one disease is a lengthy, inefficient, and costly exercise. Let us take the example of HER2 mutations that, contrary to HER2 gene amplification, occur at a low frequency of 1–2% in advanced breast cancer. There is preliminary evidence that these mutations are “actionable” using a potent irreversible tyrosine kinase inhibitor such as neratinib.8 Conducting a “confirmatory” phase II trial of around 40 patients would require the screening of 2000–4000 women with advanced breast cancer. Moreover, at the end of this exercise, the potential value of the new drug versus standard of care or in other tumor types harboring similar mutations would remain unknown. Figure 2 highlights the challenges of a registration path of such a drug in a “small” genomic segment of the breast cancer (BC) population. This genomic aberration, therefore, would be best matched with its candidate drug in the context of an “umbrella” discovery trial, where a panel of genomic aberrations would be screened and investigated in the context of several breast cancer phase II arms. Alternatively, a basket trial could examine HER2 mutations across several tumor types and investigate the activity of the TKI (tyrosine kinase inhibitor) in each of them with “early stopping rules” in case of lack of antitumor activity. These increasingly popular forms of clinical trials are described later.
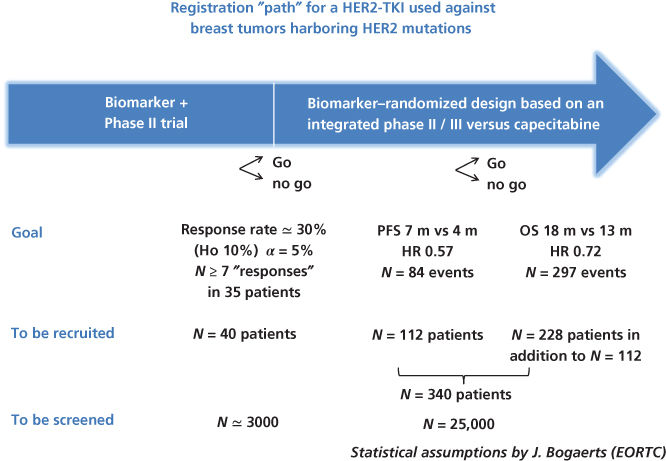
Figure 2 Schema for phase II and III clinical trials for developing a drug that inhibits a target, which is mutated in only 1–2% of cases. Although the number of patients required for the clinical trials are reasonable, the number screened is very large. It is more efficient to incorporate development of such a drug in an infrastructure of common national and international screening with regard to multiple targets and then triaging patients to clinical trials for which they are eligible.
“Basket” discovery trials include patients with advanced cancer of multiple primary sites that are resistant to standard treatment.9 A sequencing assay is used to evaluate the tumor DNA of a patient and it is determined whether an actionable aberration is present. “Actionable” means that a drug is available whose range of molecular targets overlap with the genomic alterations of the tumor in a way that suggest treatment may result in benefit for that patient. Various kinds of evidence may be used to determine whether a drug is a reasonable candidate for a given mutation. It will include biological understanding of the targets of the drug and the role of those targets in the disease. It may involve using the COSMIC (Catalog of Somatic Mutations in Cancer) database to determine whether the gene is mutated frequently enough in that histologic type to be considered a “driver” mutation. It may involve using algorithms to determine whether mutations found in a gene are predicted to alter protein function. It may also involve using preclinical data about drug activity in cell lines, tumor grafts bearing that mutation in mice, or clinical data in a different tumor type. The rules of actionability should be prospectively defined. Defining levels of evidence for actionability of a drug and a genomic aberration may help trial organizers resolve difficult binary decisions of whether there is sufficient evidence to warrant considering a drug actionable.
The term “basket trial” is often reserved for clinical trials of a single drug with multiple histologic types of patients and multiple types of mutations of either a single gene or several genes. The trials with multiple drugs involved are often called “umbrella trials.” Both types of trials are early discovery trials and attempt to identify the genomic characterization of tumors for which there is evidence of substantial anticancer activity of a drug. These positive signals should be confirmed in later expanded phase II or II/III trials.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


