Biology of Adipose Tissue
Sheila Collins
Rexford S. Ahima
Barbara B. Kahn
EVOLVING CONCEPTS ABOUT ADIPOSE TISSUE PHYSIOLOGY
From an evolutionary perspective, the biology of adipocytes begins with the strategies developed by organisms over time to cope with irregular and unpredictable supplies of nutrients. The need for such a mechanism became especially acute in omnivorous mammals and was probably a key survival factor. As Homo sapiens developed complex societal rituals that revolved around shared meals and as social status was assessed by the acquisition of resources, to be well endowed with a layer of fat was a clear sign of success, wealth, and social dominance. However, in more recent times, particularly in industrialized societies over the last 40 years, it has been more popular to possess the least amount of body fat. This modern idealism of slimness represents the new prototype of the species, glamorized by fashion and art. Simultaneously, in the medical community, there is also a greater emphasis on a leaner physique because of the growing alarm over the epidemic of overweight and obesity in adults and even children (see Chapter 31 for a detailed definition and discussion of obesity). The most recent statements from the Surgeon General of the United States and the World Health Organization emphasize the need for the general public to lose weight (specifically to reduce percentage of body fat) because of the established associations between excess body fat and increased risk for a host of serious medical complications (1) (http://www.cdc.gov/nccdphp/dnpa/obesity/trend/index.htm), including type 2 diabetes and cardiovascular disease, to name just two. It is also recognized that extreme deficiencies in body fat, such as lipodystrophy, can be as detrimental as excess fat in terms of diabetes risk (2). It is therefore important to understand the development and metabolic functions of adipose tissue and how these processes are regulated.
Until the 1990s, adipose tissue had largely been considered an inert storage depot for excess metabolic fuel. Accumulation of excess calories as triglycerides in adipose tissue is driven largely by insulin, and subsequent access to this stored fuel is gated by the β-adrenergic catecholamine receptors and their ability to stimulate lipolysis. The discovery of leptin as an adipose-derived hormone that can “report” on the status of these energy reserves to other organs of the body, including the central nervous system, gave us a new perspective on the biology of adipose tissue (3). The ensuing years have brought a deeper appreciation of the secretion by adipose tissue of a fairly large number of cytokines and growth factors that may play significant roles in insulin resistance, cell differentiation, and growth (Fig. 13.1). We will review aspects of the biology of adipocytes ranging from general anatomy and cellular and molecular biology to exciting new developments that support the concept that adipose tissue is an endocrine organ and that the adipocyte is a critical player in the regulation of insulin sensitivity as a result of its ability to secrete fatty acids, leptin, various cytokines, and other novel peptides.
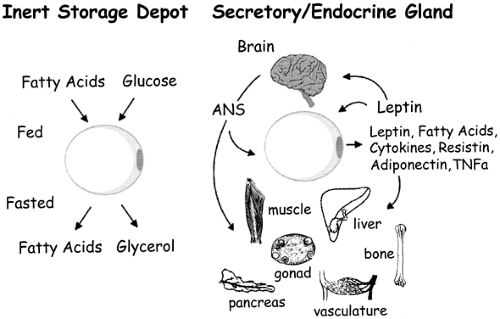 Figure 13.1. Evolving view of the biologic function of the adipocyte. Previously, adipocytes were considered to be inert storage depots releasing fuel as fatty acids and glycerol in times of fasting or starvation. More recently, it has become clear that adipocytes are endocrine glands that secrete important hormones, cytokines, vasoactive substances, and other peptides, with far-reaching effects on other organs and tissues, including the brain. TNF, tumor necrosis factor; ANS, autonomic nervous system; ACRP30, adipocyte complement-related protein–30 kDa (adiponectin). (From Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000;106:473–481, with permission from the American Society for Clinical Investigation.) |
ADIPOSE TISSUE: STRUCTURAL FEATURES AND ANATOMIC DISTRIBUTION
Adipose tissue is present in every mammal and is critical to the maintenance of energy balance. Until the past few years, adipose tissue had been considered a type of loose connective tissue composed of lipid-filled cells (adipocytes) surrounded by a matrix of collagen fibers and a supporting blood supply. However, the current view of adipose tissue is that of a bona fide organ system with distinct tissue types mediating specific functions [e.g., homeostasis of energy reserves, hormone secretion, and immune regulation (4)]. Adipose tissue has been classified into two major forms on the basis of gross appearance, cell type-specific gene expression, and predominant type of adipocytes (5): white adipose tissue and brown adipose tissue.
White Adipose Tissue
White adipose tissue (WAT) is named for its white/yellowish color. It contains adipocytes with a single large lipid inclusion (termed unilocular), resulting in displacement of the cytoplasm, flattening of the nucleus to the plasmalemma, and appearance of signet ring-shaped adipocytes. After histologic processing with xylene or other fat solvents, WAT sections lose lipid, such that tissue resembles a meshwork of polygonal or ovoid profiles of variable sizes (20 to 200 μm diameter) when seen under the light microscope (Fig. 13.2). WAT is considered to be less well vascularized than brown adipose tissue (BAT). However, capillaries often are observed at the junctions where adipocytes meet. Heavier connective tissue septa separate WAT depots into lobules of various sizes. Special stains have revealed that the connective tissue surrounding white adipocytes contains precursor cells, fibroblasts, immune cells, reticular fibers, and unmyelinated nerves (6). The nerve fibers have been found to be immunoreactive for tyrosine hydroxylase, calcitonin gene-regulated peptide (CGRP), neuropeptide Y, and substance P (7). The postganglionic tyrosine hydroxylase-positive sympathetic nerves terminate primarily on arterioles and, when stimulated, cause vasoconstriction. In support of earlier anatomic and histologic observations, there is good evidence of a direct sympathetic innervation of white adipocytes originating from the central nervous system (8,9). Electron microscopic examination of white adipocytes shows that they are surrounded by a basal lamina. The cytoplasm is compressed into a thin rim containing a small Golgi complex, endoplasmic reticulum, free ribosomes, and filaments (9,10,11). Pleomorphic mitochondria are located close to the nucleus. The large lipid inclusion is not surrounded by a membrane but is heavily decorated with a protein called perilipin (12). The lipid droplet is also devoid of organelles.
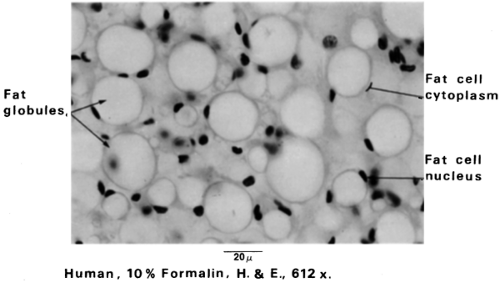 Figure 13.2. White fat cells. The cytoplasm appears as a thin rim at the periphery of the cell. Stored fat is the predominant component of the cell. The nucleus is flattened in the cytoplasm, permitting maximum storage of fat globules. The fat cell lipid is in the form of a single droplet, and these cells are described as unilocular. (From Bergman RA, Afif AK, Heidger PM. Atlas of microscopic anatomy, 2nd ed. Philadelphia: WB Saunders, 1974:53 [see color plate], with permission.) |
WAT is the predominant type of adipose tissue in adult mammals. In healthy humans it accounts for 15% to 20% of body weight in men and 20% to 25% in women. The majority of WAT is organized into a continuous layer in the subcutaneous tissue called the panniculus adiposus. Subcutaneous WAT is especially abundant in the lower abdomen and buttocks, particularly in females. Other WAT depots are located in the omentum, breast, mesentery, and retroperitoneal/perirenal regions in humans, some of which can become pathologically large. WAT also accumulates in the axilla, pericardium, bone marrow, orbits, hands, and soles of the feet and functions as a cushion in these latter regions. In rodents, subcutaneous WAT accumulates around the scapular, axillary, and inguinal regions. Intraabdominal depots are located in the retroperitoneal area and in
the mesenteric and perigonadal regions. The sexual dimorphism of body contour in humans is accounted for in large measure by differences in subcutaneous WAT (13). In addition to its abundance in the breasts, WAT is more abundant in the hypodermis of the lower abdomen, buttocks, and thighs in women than in men. These differences are influenced in part by sex hormones, as the female (gynoid) pattern of fat distribution tends to be altered to a central (android) location at menopause. Excessive central adiposity (especially intraabdominal) has been linked to the “metabolic syndrome,” which includes insulin resistance, dyslipidemia, and increased risk of cardiovascular disease (14).
the mesenteric and perigonadal regions. The sexual dimorphism of body contour in humans is accounted for in large measure by differences in subcutaneous WAT (13). In addition to its abundance in the breasts, WAT is more abundant in the hypodermis of the lower abdomen, buttocks, and thighs in women than in men. These differences are influenced in part by sex hormones, as the female (gynoid) pattern of fat distribution tends to be altered to a central (android) location at menopause. Excessive central adiposity (especially intraabdominal) has been linked to the “metabolic syndrome,” which includes insulin resistance, dyslipidemia, and increased risk of cardiovascular disease (14).
In general, the proportion of WAT in healthy adults remains fairly constant over prolonged periods. However, WAT can increase markedly with obesity and decrease with anorexia nervosa, cancer, and other wasting diseases. By weight, WAT comprises between 70% and 90% lipid, 5% to 30% water, and 2% to 3% protein. Most of the lipid content is triglyceride (>90%), although small amounts of diglyceride, cholesterol, and phospholipid are present. As noted above, in all species the ability to store calories efficiently within WAT can confer enormous survival advantage against the threat of starvation. As will be discussed later, fat metabolism is dependent on energy requirements and is regulated by nutrient, neural, and hormonal signals. The postprandial increase in glucose and lipids stimulates insulin release and increases fatty acid transport and lipogenesis in adipocytes. Conversely, reductions in glucose and insulin during fasting and stimulation of the sympathetic nervous system lead to lipolysis and the release of fatty acid for use by other tissues.
The morphology of WAT is affected by nutritional status. Obesity is an increased volume of WAT due to hypertrophy and hyperplasia of adipocytes (15,16,17). Lipid accumulation is associated with the formation of numerous micropinocytotic invaginations and vesicles, which coalesce into multilocular and finally unilocular lipid inclusions (10,11). By contrast, fasting causes a reduction in the size of the lipid droplet, an irregularity of the plasmalemma associated with numerous micropinocytotic invaginations and vesicles, and a prominent smooth endoplasmic reticulum (18). Prolonged fasting causes white adipocytes to take on the appearance of spindle-shaped fibroblastlike cells containing very few lipid inclusions (19). Moreover, the amount of capillaries is increased during fasting, consistent with the reported rise in blood flow in WAT, to facilitate delivery of oxygen and transport of the hydrolyzed free fatty acids (FFAs) to other tissues (20).
In addition to providing nourishment, proteins secreted by vascular endothelium control adipocyte differentiation and maturation (21). Antiangiogenic factors, e.g., angiopoeitin and TNP-40, inhibit angiogenesis as well as adipogenesis, and deplete adipocyte lipid stores in rodents (22,23). Vascular endothelial growth factor (VEGF) is increased with visceral fat accumulation, stimulated by insulin, and decreased by weight reduction (24,25). Importantly, blockade of VEGF inhibits adipogenesis, suggesting a causal role in adipose tissue development (25). In a recent study, targeting of a novel peptide to prohibitin, a vascular marker of adipose tissue, resulted in destruction of adipose vasculature and rapid reversal of obesity in rodents (26). These findings have generated considerable interest in the use of antiangiogenic agents as treatment for obesity.
The idea that obesity is associated with chronic inflammatory response, e.g., abnormal production of acute-phase reactants, and induction of inflammatory signaling pathways, is an old one (27). Chronic low-grade inflammation had been linked to insulin resistance, diabetes, and cardiovascular disease in obese individuals, although the underlying mechanisms remained unclear until recently. There are remarkable similarities in the patterns of gene expression, activation of complement, and induction of cytokines, among T-cells, macrophages, and adipocytes. Importantly, adipocyte precursors have the capacity to be transformed into phagocytic cells. Futhermore, there is significant overlap between metabolic pathways that mediate lipid metabolism in macrophages and adipocytes. Obesity in rodents and humans triggers a progressive infiltration of the stromovascular compartment of adipose tissue by macrophages (28,29). This process appears to be mediated through production of TNF-α and other cytokines by obese adipocytes, which in turn stimulate preadipocytes and endothelial cells to produce matrix proteins, e.g., monocyte chemoattractant protein (MCP)-1. Macrophage infiltration may be further enhanced by adipocyte hormones, e.g., leptin and adiponectin, as well as oxidative injury to endothelium. A key question is how these local changes eventually lead to diabetes and cardiovascular complications of obesity. Perhaps, cytokines and other chronic inflammatory signals from obese adipose tissue directly regulate glucose metabolism in muscle, liver as well as pancreatic β-cell responses, as shown by the improvement in glucose levels in parallel with inhibition of NF-κB by salicylate (30).
Brown Adipose Tissue
The rich and varied history of BAT as an anatomically discrete type of fat includes early speculations in the 17th century that it was part of the thymus and, a century later, that it was an endocrine organ involved in blood formation or a special form of fat acting as a reservoir for certain nutrients (31). BAT is named for its “brown” color, which is derived from a rich blood supply and an enormous number of mitochondria per cell (5). In overall appearance the adipocytes in BAT are generally smaller than white adipocytes and characterized by numerous small lipid inclusions (termed multilocular) (Fig. 13.3). Unlike white adipocytes, the nucleus is eccentric but not flattened.
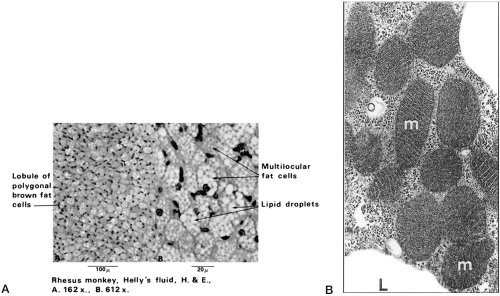 Figure 13.3. A: Brown fat is an uncommon variety of fat found in specific locations in the body. Unlike the more common white fat, brown fat cells contain a number of small lipid droplets; hence the name multilocular fat. Panel B illustrates the dense mitochondrial content of brown adipocytes. (From Bergman RA, Afifi AK, Heidger PM. Atlas of Microscopic Anatomy, 2nd ed. Philadelphia: WB Saunders, 1989: 54 [see color plate], with permission.) |
Only in 1961 was the true function of BAT realized, when it was proposed to be thermogenic (32,33). Since then, an immense body of work has shown that BAT is uniquely capable of responding to various environmental stimuli to generate heat from stored metabolic energy. In response to activation by the sympathetic nervous system, BAT undergoes an orchestrated hyperplastic and hypertrophic expansion, increased blood flow, and recruitment of lipid and carbohydrate fuels for oxidative metabolism (34,35). A unique and critical element of this thermogenic mechanism for dissipation of the proton gradient in brown fat mitochondria was recognized to be due to a brown fat-specific mitochondrial uncoupling protein (UCP) (36), also called thermogenin (37). As illustrated in Figure 13.4 and discussed in greater detail by Ricquier and Bouillaud (38), this mitochondrial protein, now known as UCP1, allows controlled proton leakage across the mitochondrial inner membrane for the purpose of generating heat at the expense of respiration-coupled ATP production. This uncoupling activity in brown fat mitochondria is “activated” by FFAs that are released as a result of catecholamine-stimulated lipolysis.
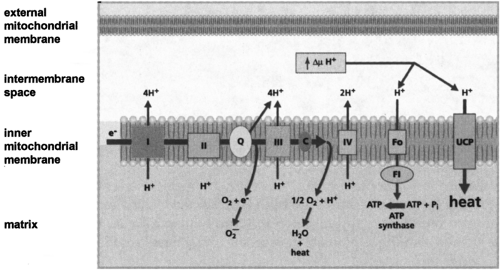 Figure 13.4. Transport of electrons through the respiratory chain complexes (I–IV) is associated with pumping of protons from the mitochondrial matrix into the intermembrane space, creating an electrochemical gradient (ΔμH+) for adenosine triphosphate (ATP) synthesis by the ATP synthase (F0–F1). An uncoupling protein (UCP) provides an alternative route for protons to reenter the matrix, thereby uncoupling the oxidation of fuel from ATP production. (Drawing courtesy of Dr. Antonio Vidal-Puig.) |
Developmentally, BAT is most abundant in newborn mammals and is principally involved in heat production (5,39). In the human fetus, BAT is located in the dorsal cervical, axillary, suprailiac, and perirenal regions. Smaller amounts are present in the anterior mediastinum, interscapular, intercostal, and retropubic regions. By contrast, BAT is very prominent in the interscapular and axillary areas in mice and rats, with lesser deposits in the dorsal midline of the thorax and abdomen. Light microscopy examination shows that these BAT depots that
reside along the midline are partitioned into lobules by dense connective tissue containing numerous blood vessels and nerves. For example, the prominent interscapular BAT in the rat receives two arteries and veins and has abundant capillaries (9). The nerve supply consists of large myelinated fibers, as well as small unmyelinated fibers that stain intensely for neuropeptide Y and tyrosine hydroxylase (9). The latter, which are postganglionic sympathetic fibers, form a periarterial plexus and also innervate brown adipocytes directly. In large mammals such as dogs and primates (including humans), homogenous depots of BAT decline with age beyond infancy but, as in rodents, brown adipocytes can be detected interspersed within typical “white” adipose depots throughout adulthood [reviewed in reference (40)]. Moreover, cold challenge or treatment with a β3-adrenergic receptor (β3AR) agonist further provokes the elaboration of such brown adipocytes in all of these animals. Humans with pheochromocytoma exhibit large amounts of BAT as a result of chronic catecholamine stimulation (41).
reside along the midline are partitioned into lobules by dense connective tissue containing numerous blood vessels and nerves. For example, the prominent interscapular BAT in the rat receives two arteries and veins and has abundant capillaries (9). The nerve supply consists of large myelinated fibers, as well as small unmyelinated fibers that stain intensely for neuropeptide Y and tyrosine hydroxylase (9). The latter, which are postganglionic sympathetic fibers, form a periarterial plexus and also innervate brown adipocytes directly. In large mammals such as dogs and primates (including humans), homogenous depots of BAT decline with age beyond infancy but, as in rodents, brown adipocytes can be detected interspersed within typical “white” adipose depots throughout adulthood [reviewed in reference (40)]. Moreover, cold challenge or treatment with a β3-adrenergic receptor (β3AR) agonist further provokes the elaboration of such brown adipocytes in all of these animals. Humans with pheochromocytoma exhibit large amounts of BAT as a result of chronic catecholamine stimulation (41).
Development of Adipose Tissue
The origin of adipose tissue was a matter of controversy for a number of years (42,43). Early histologists debated whether adipose tissue was a distinct organ or merely a specialized loose connective tissue with lipid-filled fibroblasts. The current view of adipose tissue as a distinct organ was based on studies showing that adipocyte precursors were derived from mesenchyme and that the blood supply of the so-called primitive fat organ was distinct from the surrounding connective tissue. In humans and most mammals, adipose tissue development begins at midgestation. In rats and mice, WAT does not develop until the perinatal period, but established BAT depots are present in the late stages of gestation. Adipose depots arise from “undifferentiated” cells clustered along blood vessels. Adipocyte precursor cells lack lipid droplets and are associated with a rich supply of proliferating capillaries. The transformation of preadipocytes into white adipocytes is characterized morphologically by the
accumulation of small lipid droplets that eventually coalesce into a single large droplet.
accumulation of small lipid droplets that eventually coalesce into a single large droplet.
The concept that preadipocytes exist within adipose tissue, even into adulthood, and that they can differentiate into adipocytes under appropriate hormonal stimulation is supported by studies showing that cells isolated by collagenase digestion from the stromal-vascular compartment of fat pads from many sources, including humans, can differentiate into adipocytes when cultured in vitro in media enriched with insulin and other serum factors [reviewed in reference (44)]. In addition, studies of obesity in various strains of mice indicate that adipocyte hyperplasia can exist into adulthood (45). The ability of adipocytes to replenish a depot following ablation by gene targeting also supports this idea (46). Our understanding of the biochemistry and genetic program controlling adipocyte differentiation has also benefited enormously from studies of immortalized mouse preadipocyte cell lines such as 3T3-L1 and 3T3-F442A (47). These cells are propagated as fibroblastlike preadipocytes that are capable of differentiating into mature adipocytes under appropriate hormonal stimulation and that for the most part express the same genes as white adipocytes in vivo. As a result of extensive studies in these and similar adipogenic cell lines, we know that the adipocyte differentiation program proceeds through a series of well-characterized stages: (a) preconfluent, (b) growth arrest/confluence, (c) clonal expansion, and (d) terminal differentiation. Each stage involves a coordinated expression of transcription factors to culminate in the expression of specific genes for lipid metabolism, hormones, cytokines, and other adipocyte products (44,48).
MOLECULAR EVENTS IN PREADIPOCYTE COMMITMENT AND DIFFERENTIATION
The quest to define the critical required factors and/or temporally specific events in the commitment and differentiation of a given cell type has long fascinated biologists and has driven the field of molecular biology. This line of investigation in general has benefited from the efforts to understand the molecular basis of adipogenesis. The adipogenic cell lines of Green and colleagues (36) provided the technical basis from which to isolate adipocyte-specific genes and identify their regulatory factors. As a result of such approaches, a number of key transcription factors are now known to promote adipogenesis. They include the family of CCAAT/enhancer binding proteins (C/EBP), peroxisome proliferator-activated receptor-γ (PPAR-γ), and the sterol regulatory element binding protein (SREBP) family (SREBP1c, also known as ADD1). As shown in Figure 13.5, during adipocyte commitment and differentiation, the expression of the C/EBPs and PPAR-γ2 occurs in cascade fashion. A large number of studies in cell culture models and in genetically modified animals clearly show that agents or manipulations that interfere with the expression of these factors can inhibit differentiation or result in an incomplete state of differentiation. The genes and signaling pathways that commit cells to the preadipocyte lineage have been less well studied but are now an active area of investigation. In addition, although some of these cell culture models can appear morphologically to differentiate into adipocytes (49), it is now appreciated that in some cases the failure to appropriately express certain transcription factors results in the absence of gene products responsible for critical metabolic activities (50,51,52).
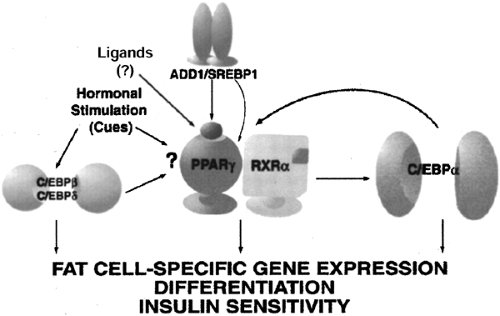 Figure 13.5. The transcriptional control of adipogenesis involves the activation of a variety of transcription factors. These proteins are expressed in cascade fashion, in which C/EBPβ and C/EBPδ are among the earliest seen. These two proteins promote the expression of peroxisome proliferator-activator receptor-γ (PPAR-γ), which in turn activates C/EBPα. C/EBPα feeds back on PPAR-γ to maintain a differentiated state. ADD1/ SREBP1 can activate PPAR-γ by inducing its expression, as well as by contributing to the production of an endogenous PPAR-γ ligand. All these factors contribute to the expression of genes that characterize the terminally differentiated phenotype. (From Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 2000;16:145–171, with permission from the Annual Review of Cellular and Developmental Biology, © 2000 by Annual Review.) |
The PPAR-γ isoform PPAR-γ2 is expressed at high levels in adipose tissue (53,54). Its identification was based on a search for transcription factor(s) implicated in the expression of adipocyte-specific genes (55,56,57). Fibroblasts such as NIH-3T3 that are genetically engineered to express PPAR-γ2 can be coaxed to differentiate into “adipocytes” by the definition of visual proof of lipid accumulation and the expression of genes related to triglyceride synthesis and storage (53). In agreement with a key role for PPAR-γ2, its failure to be expressed during development prevents adipogenesis (58,59), and deletion of the PPARγ gene from mature adipocytes in vivo results in depletion of cells from the depot and decreased adipose mass (46). Provision of PPAR-γ2 by retroviral infection to PPAR-γ-deficient cells fully restores the capacity for differentiation (60,61).
While these results support the primacy of PPAR-γ for adipogenesis, several issues remain unresolved. An “endogenous ligand” for PPAR-γ remains undefined. At present there are two views of this situation. One is that a single, specific, high-affinity ligand is produced in target tissues. A second, based on crystallographic data (62) and comparisons of fatty acid binding and activation, is that the ligand-binding pocket of PPAR-γ can accommodate a variety of ligands, leading to the proposition that the receptor may serve as a general sensor of fatty acid milieu: the so-called PUFA receptor (for p oly u nsaturated f atty a cids), with no unique fatty acid ligand. In some sense this latter idea is akin to the ability of the cytochrome P450 family to
accommodate a variety of hydrophobic substrates in the active site of the enzyme, hydroxylating them with turnover numbers that are relatively slow for highly specific enzymes, but nevertheless successfully accomplish the goal of identifying and eliminating xenobiotics (63).
accommodate a variety of hydrophobic substrates in the active site of the enzyme, hydroxylating them with turnover numbers that are relatively slow for highly specific enzymes, but nevertheless successfully accomplish the goal of identifying and eliminating xenobiotics (63).
In addition to the endogenous ligand issue, it is clear not only that PPAR-γ is required but also that C/EBPα is critical to the expression of the full complement of genes that define the mature adipocyte phenotype. For example, in NIH-3T3 fibroblasts engineered to express PPAR-γ2 and differentiated into adipocytes (53), even in this so-called differentiated state, these cells lack C/EBPα. As a result, they also lack certain defining features of the adipocyte, such as insulin-stimulated glucose uptake (50,51) and expression of the adipocyte-specific β3AR (52). These metabolic anomalies are corrected following reintroduction of C/EBPα, confirming the need for C/EBPα in the mature adipocyte, at least for specific adipocyte genes and functions. Interestingly, absence of C/EBPα from adipose tissue in mice prevents the appearance of white, but not brown, fat (64).
Although these models have provided a wealth of information about adipocyte differentiation, it is true that most of these conclusions result from immortalized cell lines grown in isolation from other constituents of the adipose organ. Global targeted disruption of PPAR-γ2 in mice results in embryonic lethality (58,65,66). Consequently, studies of adipogenesis in PPAR-γ-deficient cells were performed in cultured cells derived from these animals. [However, as already noted, mice with adipose-specific “knockout” of PPARγ are viable, lack adipose tissue, and are insulin-resistant (59,67).] In addition, as discussed later in this chapter, the majority of these cell models in culture differentiate into white adipocytes. There have been very few available cell-culture models that differentiate into brown adipocytes, and most do not fully recapitulate the complete characteristics of brown adipocytes in vivo. Over the last decade, several immortalized brown adipocyte cell lines have been generated (68,69,70,71), providing the opportunity to advance molecular understanding of brown adipocyte differentiation and gene regulation. Several immortalized brown adipocyte cell lines were developed by a strategy called targeted transgenic tumorigenesis. Tissue-specific promoters were used to engineer SV40 T-antigen or oncogenic mutants of p53 to produce brown adipose tumors, from which cell lines were developed. Initially, the characteristics of these cell lines were, in large measure, quite representative of the brown adipocyte in vivo, but it is interesting that many of these cells have evolved with continuous passage in culture to express fewer and fewer features of the brown adipocyte: loss of expression of UCP1, depressed expression of other markers of mitochondria, and loss of β3AR expression [(69,71) and S. Collins and K. W. Daniel, unpublished observations, 1996–1999].
In other cases these brown adipocyte models also lack expression of certain brown adipocyte genes (68,72), or simply have not yet been fully characterized. One theory concerning the loss of phenotypic validity with culture passage is that the more differentiated cells in the culture, expressing UCP1, tend to be at a proliferative disadvantage and over time are lost from the population. In addition, murine preadipocyte cultures do not address the contribution of the connective tissue matrix to the maturation of adipose tissue. Finally, among the various adipose depots in vivo, there are significant differences in metabolic characteristics and expression of certain genes that presently cannot be replicated in cell culture. The reader is referred to recent reviews that provide additional details (44,73).
MOLECULAR FEATURES OF WHITE VERSUS BROWN ADIPOCYTES
Despite the wealth of knowledge that has accrued over the past 25 years about the molecular events that set in motion and maintain the phenotype of the differentiated adipocyte, our understanding of the genetic programs that distinguish white from brown adipocytes is still far from complete. Historically, the agreement about the existence of BAT in humans and the importance of brown adipocyte thermogenesis has had a very checkered past. As discussed above, it is clear that a discrete adipose depot of homogeneous brown adipocytes exists at birth but does not remain in adult humans. Nevertheless, one can also readily find “brown adipocytes” in adults, as defined by morphologic and UCP1 histochemical criteria, scattered among white adipocytes in various “white adipose” depots, including perigonadal, perirenal, and pericardial, albeit they appear to be a small percentage of total adipocytes (Fig. 13.6). These BAT depots can undergo significant hypertrophy in adult humans under conditions of chronic catecholamine stimulation, as in pheochromocytoma (41). Similarly, in small rodents that retain bona fide BAT depots in adulthood, one finds scattered brown adipocytes in intraabdominal and intrathoracic white adipose depots, but rarely in subcutaneous fat. Part of the difficulty in establishing the extent to which adult humans possess brown adipocytes is that most biopsies are collected from subcutaneous sites. Since this is not the location of brown adipocytes at birth, estimates of residual brown adipocytes in humans, are, indeed, unknown. Thus, several important unanswered questions pertaining to the issue of brown adipocytes in adult humans remain.
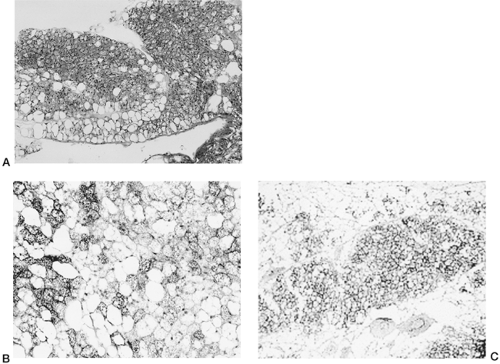 Figure 13.6. Brown adipocytes within a strip of human adipose tissue from the mediastinum. Hematoxylin and eosin stain. Courtesy of Dr. Laura Hale, Duke University Medical Center.B: Section of retroperitoneal adipose tissue from an A/J mouse following treatment with the β3-adipocyte receptor agonist CL316,243. Brown adipocytes visualized by immunostaining with antisera to uncoupling protein 1 (UCP1). From S. Collins and L. P. Kozak, unpublished observations. C: Section of inguinal adipose tissue from an AXB8 mouse after being housed at 5ºC for 7 days, showing patches of brown adipocytes as visualized by immunostaining with antisera to UCP1. (From Guerra C, Koza RA, Yamashita H, et al. Emergence of brown adipocytes in white fat in mice is under genetic control: effects of body weight and adiposity. J Clin Invest 1998;102:412–420, with permission from the American Society for Clinical Investigation.) |
First, we need accurate information about the number of brown adipocytes in adult humans. Second, even after we gain an accurate assessment of the number and locations of these scattered brown adipocytes in adult humans, we still must assess whether they are thermogenically active. Finally, we must determine whether these cells can be recruited in greater numbers in response to pharmacologic agents that behave as “thermogenic” drugs in rodent models (discussed in next section).
The unique morphologic differences between brown and white adipocytes (as discussed in an earlier section) are the result of differential gene expression during development and reflect the opposing metabolic functions of these cells: storage of caloric energy versus oxidation of caloric energy as heat. One of the most interesting questions in the field of adipocyte development is how and what molecular “decisions” are made in the mesenchymal precursor cells that give rise to white versus brown adipocytes. As discussed above, our understanding of the molecular events that drive the differentiation of white adipocytes has benefited enormously from the availability of clonal, immortalized cell lines that developmentally and phenotypically recapitulate white adipocytes.
The tissue-specific molecular markers of white and brown adipocytes have traditionally been the fatty acid-binding protein aP2 (also called 442) (55,74) and UCP1 (27), respectively. While aP2 is expressed in both white and brown adipocytes, UCP1 has been the unequivocal defining feature of brown adipocytes. However, for UCP1 to be expressed in such a highly cell type-specific manner, one would expect that other factors in brown adipocytes are required to initiate transcription of the UCP1 gene. Thus, we still do not know whether there are factors specifically expressed in brown adipocytes that are themselves tissue-specific or whether they comprise a set of gene products that are uniquely and coordinately expressed as a result of hormonal cues. Some candidate molecules that are differentially
expressed in brown versus white adipocytes are PPAR-γ-coactivator-1 (PGC1α) and retinoid orphan receptor-γ (RORγ). In particular, PGC1α appears to be an important coordinator and regulator of genes involved in oxidative metabolism (75). For example, when PGC1α is expressed ectopically in white adipocytes, they appear to be “converted” to brown adipocytes where the cells now express UCP1 as well as a panoply of genes required for the genesis of a mitochondrion (75,76). At least in rodent brown adipocytes, the participation of PGC1α also appears to be critical for catecholamine-stimulated expression of UCP1 (77).
expressed in brown versus white adipocytes are PPAR-γ-coactivator-1 (PGC1α) and retinoid orphan receptor-γ (RORγ). In particular, PGC1α appears to be an important coordinator and regulator of genes involved in oxidative metabolism (75). For example, when PGC1α is expressed ectopically in white adipocytes, they appear to be “converted” to brown adipocytes where the cells now express UCP1 as well as a panoply of genes required for the genesis of a mitochondrion (75,76). At least in rodent brown adipocytes, the participation of PGC1α also appears to be critical for catecholamine-stimulated expression of UCP1 (77).
IMPORTANT ADIPOCYTE METABOLIC ACTIVITIES AND THEIR REGULATION
As befitting the role of adipose tissue as a “bank” for the deposition and retrieval of stored metabolic energy, it is of paramount importance to understand the hormone systems that control its metabolic processes for whole-body energy homeostasis and for interpreting the malfunctioning of these systems in diseases such as in type 2 diabetes. In this section we review results from several fronts that establish the adipocyte as an endocrine organ that secretes factors that report to other organ systems about the energy reserve status of the organism. One of the pivotal discoveries in this regard that revolutionized our thinking about the overall function of adipose tissue was the identification of the adipocyte-derived hormone leptin. In addition, since the association of obesity with type 2 diabetes is a well-known clinical phenomenon and since obesity has been documented in many instances to be a major contributing factor to insulin resistance, we also discuss obesity and insulin resistance in terms of adipocyte biology. We integrate this new information into current views of the mechanisms by which insulin, catecholamines, and certain cytokines control storage and release of lipid in the adipocyte.
Leptin: An Adipocyte Hormone Regulating Food Intake and Metabolism
Humans and other mammals have an extraordinary ability to match food intake to energy expenditure over long periods so that body weight and adiposity are maintained at near-constant levels. This homeostatic balance is remarkable given the infinite variety of day-to-day physical activity and the great variation in dietary selections and timing of their intake. On the basis of this observation, Kennedy (78) proposed that a signal emanating from adipose tissue informed the brain about the status of
energy reserves, leading to alterations in feeding behavior and energy expenditure in an attempt to maintain energy balance. The concept of an “adipostat” was further supported by results of animal experiments in which food deprivation or surgical lipectomy resulted in compensatory hyperphagia and, conversely, acute forced overfeeding led to a reduction in voluntary ingestion until body weight was restored to the previous level.
energy reserves, leading to alterations in feeding behavior and energy expenditure in an attempt to maintain energy balance. The concept of an “adipostat” was further supported by results of animal experiments in which food deprivation or surgical lipectomy resulted in compensatory hyperphagia and, conversely, acute forced overfeeding led to a reduction in voluntary ingestion until body weight was restored to the previous level.
Despite considerable effort, the identity of a circulating factor linking adiposity to energy homeostasis remained elusive until technical advances in genetics and computational power were made. These developments, combined with the extensive biochemical, physiologic, and genetic studies of the ob/ob (obese) and db/db (diabetes) mutant strains of mice, led to the isolation in the mid-1990s of the genes for these loci. Friedman and colleagues (79) determined that the obese gene encodes a protein with a relative molecular mass of 16 kDa that is expressed mainly in adipose tissue and circulates in plasma. It has a helical structure similar to that of cytokines and is highly conserved among species. They named this adipocyte-secreted hormone “leptin” (from the Greek leptos meaning thin) because it decreased body weight when injected in mice [reviewed in reference (79)]. Moreover, Friedman and others also showed that, as originally postulated by Coleman and colleagues (80,81), the diabetes locus encodes the receptor for leptin (82,83). Together these two discoveries were a major step forward in establishing the existence of an “adipostat,” and the work led to a deeper appreciation of the endocrine role of adipose tissue (Fig. 13.1). As will be discussed later, this endocrine status of adipose tissue has expanded beyond what was initially implicated from the isolation of leptin.
REGULATION OF LEPTIN PRODUCTION AND KINETICS
There is a strong positive correlation between percent body fat, plasma concentrations of leptin, and expression of the leptin gene in adipose tissue. Moreover, leptin levels are dependent on nutritional status. They are higher in obese individuals and increase with overfeeding. By contrast, leptin levels are lower in lean individuals and decrease during fasting. The reduction in leptin expression and its circulating levels during fasting is rapid and precedes weight loss, suggesting that leptin might serve as a signal for impending energy depletion. Plasma leptin levels are related to the feeding cycle. In humans, concentrations of leptin appear to peak at night and reach a nadir in the morning. By contrast, in rodents concentrations of leptin reach a nadir during the light cycle and peak after the onset of feeding during the dark cycle, consistent with their nocturnal lifestyle. Various studies have suggested that the nutritional regulation of leptin is mediated at least in part by insulin (84), since the postprandial increase in insulin is temporally associated with increased expression of leptin messenger RNA (mRNA). Conversely, leptin decreases in response to insulin deficiency in humans and rodents. Although insulin stimulates the synthesis of leptin in adipocyte culture, and circulating levels of leptin in vivo, this effect appears to be due to insulin-stimulated glucose uptake and its metabolism (85,86) rather than to an effect of insulin signal transduction mechanisms per se on gene expression. This concept of insulin regulation is further complicated by the recent observation that mice lacking insulin receptors in fat have reduced fat mass but inappropriately high levels of leptin (87).
Several other hormonal or environmental factors have been noted to modulate leptin levels (84). For example, increases in circulating levels of leptin have been reported in response to acute infection, inflammatory cytokines, and chronic glucocorticoid and estrogen exposure. Although still largely empiric observations, the latter is postulated to be responsible, at least in part, for the higher concentration of leptin in females than in males. Higher expression of leptin in subcutaneous adipose tissue also is thought to play a role. Factors that have been associated with decreases in leptin levels include testosterone, cold exposure, adrenergic stimulation, growth hormone, thyroid hormone, melatonin, smoking, and thiazolidinediones.
ROLE OF LEPTIN IN FEEDING BEHAVIOR AND METABOLISM
At the time of its discovery, there was wide speculation that leptin was the long sought “satiety” and “antiobesity” factor described several decades ago [reviewed in references (35,36,38)]. This view was based on the following observations. First, ob/ob mice with a genetic deficiency in leptin due to a truncated gene product develop early-onset obesity, impaired thermogenesis, and hyperphagia. Similar abnormalities were observed in db/db mice and fa/fa rats with mutations in the leptin receptor, suggesting that leptin was involved in the negative feedback regulation of feeding and adiposity. Second, administration of leptin peripherally (and, more potently, by the intracerebroventricular route) prevented weight gain in ob/ob mice (and to a lesser extent in wild-type mice) through inhibition of food intake and increased energy expenditure, in agreement with the notion that leptin was the missing antiobesity factor in these mice (88,89,90).
Leptin-mediated weight loss also may involve effects on lipid metabolism and adipocyte survival, as peripheral or intracerebroventricular injection of leptin has been shown to increase fat oxidation and to promote adipocyte apoptosis [discussed in reference (79)]. The increase in energy expenditure following the administration of leptin is mediated by activation of the central sympathetic pathway (91,92) and the predicted downstream stimulation of brown fat-specific uncoupling proteins. Since the receptors and neuroendocrine cells controlling food intake and energy expenditure are situated in the hypothalamus and certain other brain regions (93,94,95), it must be understood how leptin, produced by adipocytes and released into the general circulation, arrives at these privileged sites within the central nervous system. There is general agreement from the current literature that leptin is delivered from the circulation into the central nervous system by a saturable, facilitated transport mechanism. The so-called long form of the leptin receptor (Ob-Rb), which mediates activation of the JAK-STAT (J anus K inase— S ignal t ransducer and a ctivator of t ranscription) signal transduction pathway, has been colocalized with key neuropeptides and neurotransmitters in these brain regions involved in feeding behavior and energy balance [reviewed in reference (96,97,98)]. Recent data suggest that in addition to the Jak Stat pathway, leptin also activates the PI3 Kinase pathway and this may play a role in some of its metabolic effects (99,100). In addition, in skeletal muscle leptin activates the AMP-activated protein kinase (AMPK), and this is necessary for its effect on acetyl CoA carboxylase and stimulation of fatty acid oxidation (100a). In the hypothalamus, leptin and other anorexigenic signals inhibit AMPK activity (100b,100c), and this inhibition is necessary for leptin’s effect on food intake and body weight (100c).
LEPTIN AS AN ANTIOBESITY HORMONE
The critical role of leptin in regulation of appetite and body weight has been demonstrated in rodents and humans with genetic leptin deficiency or leptin-receptor mutations; however, such mutations are extremely rare [see reference (101)]. The notion of leptin as an antiobesity hormone could be considered to be inconsistent with the empiric observations in both humans and rodent models that high endogenous levels of leptin do not prevent the accumulation of excess adipose tissue (102). However,
a counterpoint argument is that the manner in which hyperleptinemia is indicative of “leptin resistance” is markedly similar to hyperinsulinemia in type 2 diabetes. In the latter circumstance it cannot be argued that the phenomenon of “insulin resistance” thus negates the role of insulin in controlling glucose homeostasis. Mechanisms underlying leptin resistance might include impairment of leptin synthesis or secretion, a decrease in transport of leptin across the blood-brain barrier, and abnormal leptin-receptor or postreceptor signaling. Data support both of these latter two possibilities. As expected, leptin treatment reverses hyperphagia, obesity, and hormonal and metabolic abnormalities when administered in ob/ob mice (88,89,90). Similarly, leptin drastically inhibits appetite, reverses obesity and neuroendocrine and immune abnormalities in the few humans with congenital leptin deficiency (103).
a counterpoint argument is that the manner in which hyperleptinemia is indicative of “leptin resistance” is markedly similar to hyperinsulinemia in type 2 diabetes. In the latter circumstance it cannot be argued that the phenomenon of “insulin resistance” thus negates the role of insulin in controlling glucose homeostasis. Mechanisms underlying leptin resistance might include impairment of leptin synthesis or secretion, a decrease in transport of leptin across the blood-brain barrier, and abnormal leptin-receptor or postreceptor signaling. Data support both of these latter two possibilities. As expected, leptin treatment reverses hyperphagia, obesity, and hormonal and metabolic abnormalities when administered in ob/ob mice (88,89,90). Similarly, leptin drastically inhibits appetite, reverses obesity and neuroendocrine and immune abnormalities in the few humans with congenital leptin deficiency (103).
Although leptin receptors are present in several tissues, studies indicate that the antiobesity action occurs primarily in the brain. Ob-Rb and downstream leptin signaling molecules, incuding neuropeptide mediators such as neuropeptide Y, agouti-related peptide, proopiomelanocortin (POMC), and cocaine and amphetamine-regulated transcript, are enriched in hypothalamic neurons that mediate feeding, thermogenesis, and hormonal regulation (98). Importantly, targeted ablation of Ob-Rb in neurons recapitulates hyperphagia, abnormal thermoregulation, morbid obesity, hyperinsulinemia, and other metabolic abnormalites in mice (104). In contrast, loss of Ob-Rb in liver has no substantial effects on body weight or hormone levels (104). Reduced leptin-stimulated phosphorylation of STAT3 in the arcuate nucleus of the hypothalamus has been associated with high-fat feeding in mice (104a,104b). To more fully investigate the contribution of STAT3-mediated leptin signaling, Myers and colleagues (105) replaced Tyr 1138 in Ob-Rb with a serine residue, lepr(S1138), which specifically disrupts the Ob-Rb-STAT3 signal. As is the case in db/db mice, lepr(S1138) homozygotes were hyperphagic and morbidly obese. However, infertility, impaired linear growth, and diabetes characteristic of db/db mice were absent in lepr(S1138) mutants. Whereas hypothalamic expression of neuropeptide Y was increased in db/db mice but not lepr(S1138) mutants, POMC, precursor of the anorectic leptin target α-MSH, was suppressed in both db/db and lepr(S1138) mice. Thus, it appears that the leptin-Ob-Rb-STAT3 signal is functionally coupled to distinct hypothalamic neuropeptides to control energy homeostasis, growth, reproduction, and glucose levels (105).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


