INTRODUCTION
SUMMARY
The consequences of atherosclerotic vascular disease are the leading cause of morbidity and mortality in the developed countries of the world and are rapidly approaching that status in the developing world. This chapter reviews the pathologic mechanisms of atherosclerotic disease development and progression, and details the interaction of these processes with the coagulation system. The earliest morphologically visible lesion of arterial atherosclerosis, the fatty streak, already is an advanced metabolic and immunologic locus that manifests as abnormalities of vascular tone, inflammation, cellular growth, and endothelial cell dysfunction. After years to decades, the lesions advance to form plaques that grow and eventually either impinge on the arterial lumen or rupture. Rupture of a vulnerable plaque is a catastrophic event that, through activation of both platelets and the coagulation cascade, triggers thrombosis, which leads to complete occlusion, and unless collateral circulation has already been established, results in tissue ischemia. Based on an increased understanding of the pathogenesis and consequences of atheromatous plaque development and progression, medical management of atherothrombotic syndromes has improved and is reviewed for the coronary, cerebrovascular, and peripheral arteries.
Acronyms and Abbreviations
ACC, American College of Cardiology; ACCP, American College of Chest Physicians; ACS, acute coronary syndrome; AHA, American Heart Association; apo, apolipoprotein; aPTT, activated partial thromboplastin time; CABG, coronary artery bypass graft; CAD, coronary artery disease; CAPRIE, Clopidogrel Versus Aspirin in Patients at Risk of Ischaemic Events; CCL, CC chemokine ligand; CK, creatine kinase; CVD, cardiovascular disease; ECG, electrocardiogram; eNOS, endothelial nitric oxide synthase; EPC, endothelial progenitor cell; EV, extracellular vesicle; HAART, highly active antiretroviral therapy; HDL, high-density lipoprotein; hsCRP, high-sensitivity C-reactive protein; IFN, interferon; Ig, immunoglobulin; IL, interleukin; LDL, low-density lipoprotein; Lp-PLA2 lipoprotein phospholipase A2; MCP, monocyte chemoattractant protein; MHC, major histocompatibility complex; MI, myocardial infarction; NO, nitric oxide; NOX, nicotinamide adenine dinucleotide phosphate oxidase; NSTEMI, non–ST-segment elevation myocardial infarction; PAD, peripheral arterial disease; PAI, plasminogen-activator inhibitor; PCI, percutaneous coronary intervention; SLE, systemic lupus erythematosus; SNP, single nucleotide polymorphisms; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TGF, transforming growth factor; Th, T helper; t-PA, tissue-type plasminogen activator; VCAM, vascular cell adhesion molecule; VLDL, very-low-density lipoprotein; VWF, von Willebrand factor.
ATHEROSCLEROSIS
Atherothrombosis describes a disease process that begins with atherosclerosis and predisposes to thrombosis in the artery. In the 1850s, Virchow1 described atherosclerosis as an inflammatory and prothrombotic process. Rokitansky, and later Duguid, posited that atherosclerotic lesions are initiated by incorporation of platelet lipids into the vessel wall (“encrustation”) following thrombosis. It was subsequently demonstrated that insudation of plasma lipoproteins is responsible for most of the lipid content of the atherosclerotic lesions. In 1913, Anitschkow noted atherosclerosis developing in rabbits fed a relatively high cholesterol diet. Although the involvement of inflammation in atherosclerosis has been known for more than 100 years, the molecular mechanisms of atherosclerotic disease initiation and progression have become clearer only in the recent past.2 It is now understood that the classical disagreement between the “lipid” hypothesis and the “inflammation” hypothesis of atherogenesis can be reconciled because there is direct linkage between cholesterol deposition and arterial inflammation in the process.3,4
Lipid accumulation in the arterial intima, termed a fatty streak, can occur in adolescents and may progress in paroxysmal fashion to a hemodynamically significant lesion causing arterial insufficiency. Autopsy studies of young soldiers and young trauma victims indicated that occult coronary atherosclerotic plaques are commonly present in healthy individuals in their teens and twenties.5,6 In addition, intracoronary ultrasonograph studies demonstrated the presence of coronary atherosclerosis in 37 percent of healthy heart donors age 20 to 29 years, 60 percent of those age 30 to 39 years, and 85 percent of those older than age 50 years.7 Several theories are espoused for this propitious condition. One well-recognized theory is the response to injury hypothesis whereby the inciting event that predisposes to atherosclerosis is injury to the endothelial lining of the artery. This hypothesis was formulated in animal studies that showed vessel narrowing and intimal thickening after endothelial denudation with angioplasty.8,9 However, human pathologic studies of early atherosclerotic plaques indicate that endothelium is structurally present but is dysfunctional. The dysfunctional state of endothelium induces abnormalities in vascular tone, inflammation, growth, and thrombosis. Atherosclerotic risk factors contribute to endothelial dysfunction and promote atherosclerosis. This section describes the mechanisms responsible for endothelial dysfunction and the impact of atherosclerotic risk factors.
Increasing age, male gender, and heredity are the major atherosclerotic cardiovascular disease risk factors that cannot be modified (Table 134–1). Abnormal lipids, smoking, improperly controlled hypertension, improperly controlled diabetes mellitus, abdominal obesity, physical inactivity, and psychosocial factors are established risk factors that can be modified, accounting for most of the risk of myocardial infarction worldwide in both sexes and at all ages.10
In addition to these traditional risk factors, newer risk factors have been recognized.11 With the use of highly active antiretroviral therapy (HAART), HIV-infected patients have demonstrated a dramatic overall increase in life expectancy.12 At the same time, HAART-treated HIV patients also have an increased risk of developing premature cardiovascular disease over time. Both HIV viral proteins and the antiretroviral drugs themselves cause endothelial dysfunction. They activate cell signaling cascades, induce oxidative stress, disturb mitochondrial function, alter gene expression, and impair lipid metabolism in vascular cells, macrophages, and adipocytes.13,14
Cardiovascular morbidity and mortality is also recognized to be exceedingly high in patients with chronic renal failure.15,16 Increased risk of premature atherosclerotic cardiovascular disease in patients on chronic hemodialysis has been known for many years, but recent studies point to an increased risk even at early stages of chronic kidney diseases. Low glomerular filtration rates and/or proteinuria are independently associated with increased rates of cardiovascular disease.17 Other factors, such as sympathetic overactivity,18 are likely to contribute to the pathophysiology of cardiac risk in these patients. Among other emerging risk factors is obstructive sleep apnea, in which treatment may improve cardiovascular outcomes.19
Cardiovascular risk factors and abnormal blood rheology are thought to result in endothelial dysfunction that predisposes the aorta and arteries to atherosclerotic plaque development, sparing the arterioles and capillaries (Fig. 134–1). Endothelial dysfunction is a term that encompasses perturbations in the diverse physiologic functions of normal arteries, including regulation of vascular tone, inflammation, growth, and preservation of blood fluidity. Lipid accumulation20 and endothelial dysfunction are intimately connected and seminal to the initiation and progression of atherosclerosis. Endothelial dysfunction occurs early in the development of plaque and is systemic in nature, afflicting vessels throughout the arterial circulation without gross evidence of atherosclerotic plaque formation. Emerging data indicate that proatherosclerotic genes are upregulated and antiatherosclerotic genes are downregulated in areas of turbulent blood flow, as seen at branch points of arteries,21 resulting in vascular adhesion molecule expression and recruitment of monocytes.22 The atherosclerotic plaque initially may expand outward rather than inward into the vessel wall, making some significant lesions difficult to visualize by angiography. The components of the mature atherosclerotic lesion include smooth muscle cells, macrophages, T lymphocytes, and calcification, in addition to accumulation of lipoproteins.23 Neutrophils and mast cells also are implicated in the atherosclerotic process.22 Later in the process, increased activity of matrix metalloproteinases in the atherosclerotic cap predisposes to plaque rupture or ulceration, resulting in tissue factor (TF) exposure and platelet adhesion, culminating in thrombus formation.24 The thrombus may undergo endogenous fibrinolysis with plaque healing or become occlusive and produce organ damage (e.g., myocardial infarction [MI]). In severe lesions, lamellar bone, presumably from endochondral calcification, may appear.25 There is evolving evidence that extracellular vesicles (EVs), also known as microparticles, are involved in the atherosclerotic process.26 The following sections describe in detail the major manifestations of endothelial dysfunction that occur early in the atherosclerotic process.
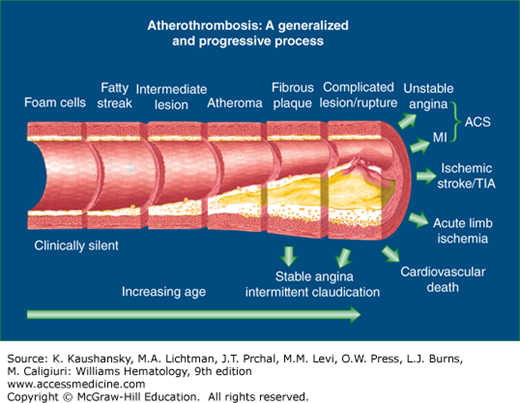
Figure 134–1.
Schematic showing the life span of the atherosclerotic plaque, beginning with the fatty streak and resulting in a thrombotic event. Cardiovascular risk factors and disturbed blood flow at branch points of vessels are thought to cause endothelial dysfunction that results in atherosclerotic plaque development in the aorta and conduit arteries. Early lipid accumulation in the intimal layer is called the fatty streak. A series of stimuli, including lipid peroxidation, are thought to signal adhesion molecule expression on the endothelium, which results in monocyte adhesion and diapedesis into the intimal space. The monocytes develop into macrophages and become sessile with accumulation of lipid (foam cells). Smooth muscle cells, primarily from the media, enter the plaque and participate in cap formation. The plaque accumulates hydroxyapatite mineral and forms calcific deposits. Matrix metalloproteinases also accumulate in the lesion and may predispose to plaque rupture or ulceration resulting in tissue factor exposure and thrombus formation. Risk factor modification favors a more stable plaque, which may have relatively less lipid accumulation and more sclerotic tissue than an unstable plaque. Severe lesions may even develop lamellar bone. ACS, acute coronary syndrome; MI, myocardial infarction; TIA, transient ischemic attack.
The importance of the endothelium in maintaining vascular tone was first recognized when endothelial cells of rabbit aorta were inadvertently removed and resulted in paradoxical vasoconstriction after administration of acetylcholine.27 The major endothelium-dependent vasodilator normally produced was found to be nitric oxide (NO), a free radical gas with multiple physiologic properties,28 including inhibition of platelet aggregation and inflammation and stimulation of angiogenesis. Numerous studies indicate that the endothelium does not vasodilate appropriately in the setting of traditional and emerging cardiovascular risk factors. Cardiovascular risk factors are thought to reduce NO availability through a variety of mechanisms, including increased oxidative stress, through generation of reactive oxygen species, and in so doing create an environment conducive to development of atherosclerosis.29 Major sources of reactive oxygen species are nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs). The catalytic subunits of the NOXs are the NOX proteins and are found in atherosclerotic lesions.30 A reduction in NO synthesis is thought to occur because of decreased availability of tetrahydrobiopterin, an essential cofactor for synthesis of NO.31 Administration of sepiapterin, a substrate for tetrahydrobiopterin, improves endothelial dysfunction.32 Also, recent evidence indicates that the transcription factor p53 and the adaptor protein Shc both play essential roles in impairing endothelium-dependent vascular relaxation.33 High cholesterol levels are thought to produce oxygen free radicals that may inactivate NO. NO synthases are the enzymes responsible for converting l-arginine to NO (Fig. 134–2) (Chap. 115). The enzyme may be perturbed by modified low-density lipoprotein (LDL), resulting in decreased NO production. Supplementation of the diet with l-arginine leads to improvement in endothelial-dependent vasodilation.34 Elevated levels of asymmetric dimethylarginine, an endogenous competitive inhibitor of NO synthase, found in patients with hypercholesterolemia and diabetes, also may result in decreased NO availability.35 Oxidized LDL is thought to increase the elaboration of asymmetric dimethylarginine by endothelial cells and decrease its degradation by the enzyme dimethylarginine dimethylaminohydrolase.36 Administration of acetylcholine to patients with elevated serum LDL37 and relatively low high-density lipoprotein (HDL)38 may result in abnormal vasoconstriction, which can be reversed with nitroglycerin (an endothelium-independent vasodilator).39 Intravenous infusion of HDL improves endothelial-mediated vasodilation through improved NO availability.40 The decreased vasodilatory capacity because of dyslipidemia may facilitate the development of coronary ischemia.
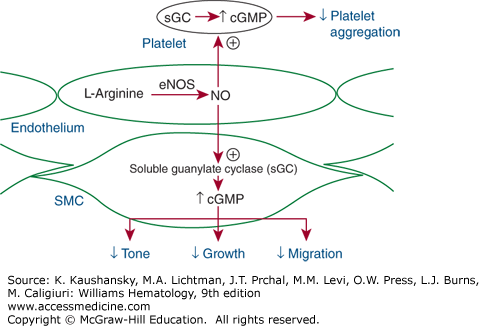
Figure 134–2.
Vascular tone depends on endothelial production and release of various vasoconstricting and vasodilating substances. The endothelial-derived vasodilators include nitric oxide (NO) and prostacyclin. NO is generated from the amino acid l-arginine by constitutive endothelial NO synthase (eNOS, or NOSIII). The enzyme is stimulated by blood flow across the endothelial surface (shear stress) or by chemical mediators, such as acetylcholine, which stimulate receptors on the endothelial surface. NO diffuses to the underlying smooth muscle cells (SMCs), where it stimulates guanylate cyclase to generate cyclic guanosine V monophosphate (cGMP), which causes smooth muscle relaxation and vasodilation. It also diffuses into blood, where it increases intraplatelet cGMP and thereby inhibits platelet adhesion and aggregation.
Impaired endothelial vasodilation is noted with advanced aging,41 when the hands are exposed acutely to cold, and during mental stress. The impairment may be mediated by increased production of endothelin, a potent vasoconstrictor.42 Sex differences are also seen in endothelial function as women in middle age tend to have more endothelial vasodilation than men at any age.43 Proinflammatory cytokines may induce formation of endothelial-derived EVs making them a surrogate marker for endothelial dysfunction44 and a biomarker for cardiovascular events.45 Infection with concomitant inflammation is associated with impaired endothelial vasodilation. For example, repeated infection with Chlamydia pneumoniae results in endothelial dysfunction via impaired NO availability.46 The combination of coronary artery disease and elevated serum levels of high-sensitivity C-reactive protein (hsCRP) is an independent predictor of abnormal endothelial vasoreactivity.47 External radiation therapy also results in endothelial dysfunction and may explain the increased risk of atherosclerosis in patients receiving mantle irradiation for Hodgkin lymphoma.48
The endothelium does not routinely interact with inflammatory cells but is poised to express adhesion molecules after stimulation with inflammatory mediators. An inflammatory response is thought to begin in the vessel wall after “invasion” of pathogenic lipoproteins.49 The presence of lipoproteins, especially oxidized LDL, results in expression of adhesion molecules such as vascular cell adhesion molecule (VCAM)-1 on the luminal surface of endothelial cells, leading to adherence of monocytes (Fig. 134–3).50 Endothelial cell expression of adhesion molecules and accumulation of monocytes can be regarded as endothelial dysfunction because these events may occur in the absence of morphologic changes in the vessel wall. Inflammation may develop without the demonstrable presence of an external microbial pathogen. It is now recognized that diseases with inflammatory component such as psoriasis and systemic lupus erythematosus (SLE) confer an increased risk for atherosclerosis and MI.51 The complex interactions of inflammation and the endothelium on the initiation and progression of atherosclerosis are reviewed in more detail in “Inflammation and Atherosclerosis” below.
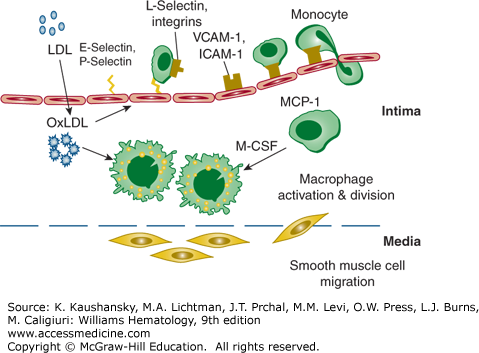
Figure 134–3.
Atherosclerotic lesion initiation is stimulated by oxidized low-density lipoprotein (OxLDL). Induction of inflammatory gene products in vascular cells is activated by the transcription factor nuclear factor-κB, which results in increased expression of cellular adhesion molecules. The adhesion molecules have specific functions for endothelial leukocyte interaction. The selectins tether and trap monocytes and other leukocytes. Vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1) mediate firm attachment of these leukocytes to the endothelial layer. OxLDL also augments expression of monocyte chemoattractant protein-1 (MCP-1 or CCL2) and macrophage colony-stimulating factor (M-CSF). MCP-1 mediates the attraction of monocytes and leukocytes and facilitates diapedesis through the endothelium into the intima. M-CSF is an important cytokine for the transformation of monocytes to macrophage foam cells. Macrophages express scavenger receptors and internalize oxidized low-density lipoprotein (LDL) during their transformation into foam cells. Smooth muscle cells migrate from the media into the intima and participate in the formation of a fibrous atheroma. (Adapted with permission from S Kinlay, AP Selwyn, P Libby: Inflammation, the endothelium, and the acute coronary syndromes. J Cardiovasc Pharmacol 32[Suppl 3]:S62, 1998.)
Normal endothelium inhibits vascular smooth muscle cell proliferation.52 The specific function of vascular smooth muscle cells in atherosclerosis is unclear. However, evidence indicates that, in early atherosclerosis, vascular smooth muscle cells contribute to the development of atheroma through production of proinflammatory mediators such as CC chemokine ligand (CCL)2 (previously termed monocyte chemoattractant protein, or MCP-1) and VCAMs. Although smooth muscle cells primarily play a role in modulating vascular tone, they also are involved in the control of extracellular matrix formation and degradation through matrix modulators such as proteases, protease inhibitors, matrix proteins, and integrins (Fig. 134–4).
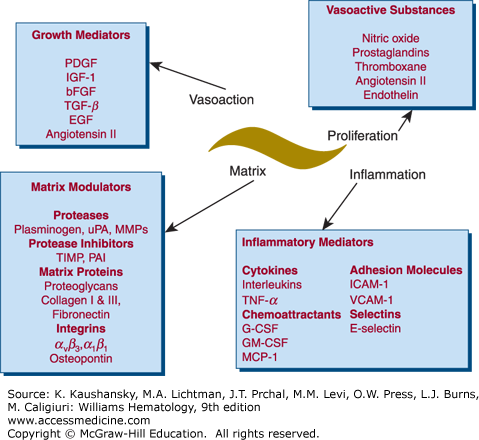
Figure 134–4.
Vascular smooth muscle cells mediate vascular proliferation, inflammation, matrix composition, and contraction. Many of these mediators have multiple functions. For example, angiotensin is a vasoconstrictor, but it also stimulates proliferation and inflammation. This is only a partial list of mediators secreted by vascular smooth muscle cells. bFGF, Basic fibroblast growth factor; EGF, epidermal growth factor; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-monocyte colony-stimulating factor; ICAM, intracellular adhesion molecule; IGF, insulin-like growth factor; MCP, monocyte chemoattractant protein; MMPs, matrix metalloproteinases; PAI, plasminogen-activator inhibitor; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β; TIMP, tissue inhibitor of metalloproteinases; TNF, tumor necrosis factor; uPA, urokinase-type plasminogen activator; VCAM, vascular cell adhesion molecule. (Adapted with permission from Dzau VJ, Braun-Dullaeus RC, Sedding DG: Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat Med 8(11):1249–1256, 2002.)
The importance of vascular smooth muscle cells in controlling the synthesis of matrix molecules is evident at the clinical level. They provide a thick, fibrous cap that promotes stability and inhibits plaque rupture and ulceration. Factor VII–activating protease, thought to play a role in coagulation and fibrinolysis, is also a potent inhibitor of vascular smooth muscle cell proliferation, and migration in vitro and local application of factor VII–activating protease (but not Marburg I variant) in animal models reduces neointima formation.53 Furthermore, it has been localized to unstable atherosclerotic plaques and may contribute to plaque instability.
Evidence indicates that vascular smooth muscle cells that undergo apoptosis, especially at the shoulder region of the plaque, may create a more unstable cap.54 Both intact vascular smooth muscle cells and fibroblasts are thought to stabilize plaques through modulation of extracellular calcification and formation of a fibrocalcific plaque.
Vascular smooth muscle cells arise primarily from the medial layer and are considered monoclonal in origin.55 Evidence also indicates that vascular smooth muscle cells may originate from the adventitia.56 The rate and timing of smooth muscle cell replication is unclear. It may occur at a constant low rate throughout the development of the atherosclerotic lesion or episodically at a higher rate. Animal studies indicate that new intimal cells may originate from outside the vessel wall from subpopulations of marrow- and non–marrow-derived circulating cells.57 Smooth muscle progenitor cells circulating in blood may contribute to the arterial remodeling that occurs after angioplasty and after bypass graft surgery.58
Vascular proliferation and inflammation are linked processes. Inflammation-induced impaired NO bioactivity contributes to vascular smooth muscle proliferation.2 Overexpression of NO synthase results in reduction of atherosclerotic or restenotic lesion formation in rabbits through both inhibition of vascular smooth muscle cell proliferation and inhibition of adhesion and chemoattractant molecule expression, with subsequent reduction of vascular mononuclear cell infiltration.59 Thus, the vascular smooth muscle cell participates in the atherosclerotic process by affecting lipoprotein retention, modulating inflammation, and controlling plaque stability through formation of the fibrous cap. Several vascular disorders involve vascular smooth muscle proliferation as the primary pathophysiologic mechanism, including in-stent restenosis, transplant vasculopathy, and vein bypass graft failure.60 The control of smooth muscle proliferation is thought to involve NR4A nuclear receptors that are expressed in atherosclerotic lesion macrophages, smooth muscle cells, and endothelial cells, and are induced by atherogenic stimuli. Inhibition of the transcriptional activity of the NR4A nuclear receptors results in enhanced smooth muscle proliferation.61 The NR4A nuclear receptors are also expressed in vein segments exposed to arterial pressure and it is postulated that they are responsible for an inhibitory feedback mechanism that occurs in activated vascular cells. Drug-eluting vascular stents that release agents such as sirolimus and paclitaxel interfere with the cell cycle and inhibit restenosis in part via decreased smooth muscle cell proliferation.62
Endothelial cells normally elaborate a number of antithrombotic substances. Some of these substances are released into blood whereas others are properties of the unactivated endothelial cell surface. These antiplatelet, anticoagulant, and profibrinolytic activities of endothelium, some of which also possess vasodilatory properties (e.g., prostacyclin, NO), act in concert to promote blood fluidity under normal circumstances. Acute activation or chronic dysfunction of endothelial cells alters the hemostatic balance, transforming them from predominantly antithrombotic to prothrombotic cells.63
To this end, endothelial cells modulate the activities of thrombin in health and disease. In the presence of intact and normally functioning endothelium, the prothrombotic actions of thrombin are quenched and the antithrombotic actions of the enzyme predominate. Thrombin binds to thrombomodulin, an integral membrane protein expressed by endothelial cells, and activates protein C (accelerated in the presence of endothelial protein C receptor, another endothelial cell protein) (Chap. 116). Activated protein C, in concert with its cofactor, protein S, has anticoagulant and profibrinolytic actions. It degrades by proteolytic digestion factors Va and VIIIa, and inactivates plasminogen-activator inhibitor (PAI)-1. Simultaneously, by binding to thrombomodulin, enzymatically active procoagulant thrombin is removed from the circulation, thereby limiting its availability to catalyze fibrin formation. Endothelial dysfunction causes loss of thrombomodulin activity from the vascular surface. In fact, increased circulating plasma levels of free (truncated) thrombomodulin represent a marker of endothelial damage. In addition to the role of thrombomodulin in clearance of circulating thrombin, the procoagulant activity of thrombin is normally blocked by endothelial cells through the action of antithrombin, which binds to heparin-like glycosaminoglycans on their luminal surface, thereby catalyzing the inactivation of thrombin by antithrombin. Like thrombomodulin, this thrombin-neutralizing action of endothelial heparan sulfate glycosaminoglycans is lost with endothelial dysfunction.
Endothelial cells do not normally express TF, but they do so upon activation by inflammatory cytokines or exposure to endothelium-activating levels of homocysteine or free thrombin. The procoagulant effects of expression of TF by dysfunctional endothelial cells are potentially compounded by the loss of TF pathway inhibitor (TFPI), which normally is synthesized by endothelial cells. Studies show that monocyte-derived EVs (or microparticles) express TF and platelet-derived EVs express phosphatidylserine and thus support coagulation complex formation.64 There is also evidence that P-selectin on platelets binds to P-selection protein ligand-1 on EVs.65
Normal endothelium is profibrinolytic. It synthesizes and releases tissue-type plasminogen activator (t-PA); it possesses binding sites for t-PA and plasminogen to provide a surface for the concentrated assembly of the fibrinolytic complex and thereby enhance local plasmin generation; and it fails to produce significant amounts of PAI-1. This profibrinolytic state is converted to an antifibrinolytic state in the presence of endothelial dysfunction. In activated or dysfunctional endothelium, PAI-1 gene expression and PAI-1 secretion are induced; simultaneously, the profibrinolytic properties of normal endothelium are lost (Chap. 135).
The antithrombotic profile of normal endothelium also manifests through the elaboration of several antiplatelet substances. NO is constitutively released into blood by normal endothelial cells and inhibits platelet adhesion and aggregation by stimulating platelet soluble guanylyl cyclase and raising intraplatelet levels of cyclic guanosine monophosphate (see Fig. 134–2).66 Physiologic flow and shear forces maintain the activity of endothelial (endothelium-derived) NO synthase (eNOS)67 under normal circumstances. Vascular cell-derived carbon monoxide, a product of heme catabolism by heme oxygenase, may have similar antiplatelet activity.68,69 Prostacyclin (prostaglandin I2) likewise is released basally by normal endothelial cells and inhibits platelet aggregation by inducing platelet adenylyl cyclase and raising intraplatelet levels of cyclic adenosine monophosphate.70
NO, carbon monoxide, and prostaglandin I2 are labile autacoids, acting only in the immediate vicinity of their release into blood from endothelial cells. An endothelial surface ecto-adenosine diphosphatase (CD39) also blocks platelet activity by metabolizing and disposing of platelet agonist adenosine diphosphate (ADP).71 In endothelial dysfunction, these various antiplatelet activities are lost, and endothelial release of von Willebrand factor (VWF) is increased, which promotes platelet adhesion. In the case of NO, oxidative stress in the microenvironment of endothelial dysfunction actually “uncouples” eNOS activity68,72 to preferentially generate superoxide over NO. Oxygen free radicals bind any remaining available NO to produce the toxic product peroxynitrite. Bioactive NO is further reduced in endothelial dysfunction by the presence of asymmetric dimethylarginine, which competes to block eNOS and limit NO production.67,73
Endothelial progenitor cells (EPCs) are heterogenous in origin and participate in endothelial cell regeneration and neovascularization of ischemic tissue. The mobilization of EPCs from the marrow is stimulated by hypoxia, cytokines such as vascular endothelial growth factor, hormones such as erythropoietin, and statin drugs, whereas mobilization is inhibited in the diabetic state.74 The role of EPCs in atherosclerosis is unclear as there are conflicting data.22 A study in apolipoprotein (apo) E–/– mice showed that there is rapid turnover of endothelial cells in atherosclerosis-prone areas and marrow derived EPCs are recruited to sites of atheroprogression.75
The endothelial response to injury manifests as a chronic inflammatory response that involves both innate and adaptive immunity.76 Innate immunity provides the first line of defense for the host and involves several cell types, most importantly macrophages and dendritic cells, which express a limited number of highly conserved sensing molecules such as scavenger receptors and toll-like receptors.76,77 Microbial infection can be detected by pathogen-associated molecular patterns, which are present in bacteria, viruses, and yeasts, but not in mammalian cells, and are recognized by the toll-like receptors.78 Ligation of a pathogen or other substances containing pathogen-associated molecular patterns (such as lipopolysaccharides, aldehyde-derivatized proteins, mannans, teichoic acids) elicits endocytosis or activation of endothelial cells (e.g., through nuclear factor-κB) that results in an inflammatory response (Chaps. 17 and 18).77,79 Proinflammatory cytokines, such as tumor necrosis factor, nuclear factor-κB and interleukin (IL)-1, magnify the innate inflammatory response.
Innate defense involves soluble factors, such as complement, which is involved in atherosclerotic lesion formation. hsCRP has been found to be an important and independent predictor for cardiovascular events.80 Natural antibodies that are generated in the absence of known antigen stimulation, mainly immunoglobulin (Ig) M, provide an immediate response against bacteria and viruses but also may be involved in atherosclerosis. For example, innate B lymphocytes, the so-called B1 cells, express a restricted set of germline–encoded antigen receptors that may bind oxidized LDL.
Compared to innate immunity, adaptive immunity is slower but more precise (Chap. 77).76 T cells can be activated by dendritic cells and macrophages, whereas most antigens cannot stimulate B cells without assistance from CD4+ T cells, which recognize the peptide–major histocompatibility complex (MHC) complexes on B cells. By genetic recombination, the number of T-cell and B-cell receptors that can be formed is almost unlimited and far exceed the number of pattern recognition receptors used by the innate immune system. Most CD4+ cells are cytokine-secreting T-helper (Th) cells and express αβ–T-cell receptors, which interact with MHC class II molecules. A smaller number of Th cells express γδ–T-cell receptors, which interact with the nonpolymorphic, nonclassic MHC molecules, CD1, which present certain antigens (particularly lipids and glycolipids). Th cells are classified according to the cytokines they secrete. Th1 cells secrete interferon (IFN)-γ and IL-2 and promote cell-mediated immunity (Chap. 78). Th2 cells secrete IL-4, IL-5, IL-10, and IL-13, and help B cells produce antibodies. CD8+ T cells are primarily cytotoxic killer cells, although they can secrete cytokines, such as tumor necrosis factor-α, IFN-γ, and lymphotoxin. Some thymus-independent antigens can activate these cells without the help of T cells. Oxidized LDL is considered such an antigen because it expresses multiple copies of oxidation-specific epitopes on a single LDL particle.
Monocyte recruitment to inflammatory foci initially involves the expression of endothelial cell selectins, which mediate monocyte rolling on the endothelium (see Fig. 134–3). The rolling phenomenon is followed by a firmer attachment to endothelial cells mediated by integrins. Perhaps the most important of these is VCAM-1, which is upregulated in cultured endothelial cells in the presence of oxidized LDL. The appearance of this molecule before the development of grossly visible atherosclerotic lesions supports oxidized LDL as an initial recruiter of macrophages. The finding of reduced atherosclerosis in VCAM-1–deficient mice further supports the important role of macrophages and VCAM-1 in the pathogenesis of atherosclerosis.81,82 Other adhesion molecules, such as P-selectin and intracellular cell adhesion molecule-1, also may be involved in monocyte adhesion at sites of lesion formation.83 The entry of monocytes into the vascular intima leads to their differentiation into resident macrophages. Here they take up cholesterol that has also accumulated in the vascular intima, thereby becoming cholesterol-engorged foam cells.84 It has been demonstrated that the accumulation of macrophage foam cells in established atherosclerotic lesions actually originates mainly from the proliferation of macrophages within the lesion rather than from the recruitment of circulating monocytes.85 Platelet-derived EVs contain and deliver the chemokine “regulated upon activation, normal T-cell expressed and secreted” (RANTES) to activate endothelium in atherosclerosis to promote attraction of monocytes.86
Lipoprotein phospholipase A2 (Lp-PLA2) is an inflammatory enzyme belonging to the large family of phospholipases that are capable of hydrolyzing the sn-2 ester bond of phospholipids of cell membranes and lipoproteins.87 This enzyme, produced by macrophages, circulates bound to LDL and in the intimal space of the artery can produce oxidized fatty acids and lysophosphatidyl choline. These molecules have a range of potentially atherogenic effects, including chemoattraction of monocytes, increased expression of adhesion molecules, and inhibition of endothelial NO production.88 Although originally designated as platelet-activating factor acetylhydrolase because of its ability to degrade platelet-activating factor, the clinical importance of this effect is not thought significant. Numerous epidemiologic studies show that Lp-PLA2 is a significant biomarker associated with cardiovascular events. The selective inhibition of Lp-PLA2 with the drug darapladib reduced development of advanced coronary atherosclerosis in diabetic and hypercholesterolemic swine.89 A phase 2 clinical study of patients with cardiovascular disease showed that sustained inhibition of plasma Lp-PLA2 activity with background of intensive atorvastatin therapy resulted in reduction in IL-6 and hsCRP after 12 weeks of darapladib 160 mg, suggesting a possible reduction in inflammatory burden.90 However, a prospective double-blind phase III trial of 15,828 patients, darapladib did not reduce the composite end point of cardiovascular death, MI, or stroke. There was a significant reduction in major coronary events and total coronary events.91
Macrophages are essential for the clearance of modified lipoproteins and the efflux of lipoprotein-derived cholesterol to HDL receptors for reverse cholesterol transport, the process by which HDL removes cholesterol from cells. Multiple lines of evidence indicate that macrophages promote lesion initiation and progression. For example, hypercholesterolemic mice become markedly resistant to atherosclerosis if they are bred to macrophage-deficient animals.92
The earliest grossly visible sign of atherosclerosis is the fatty streak, which is composed mainly of macrophage foam cells containing relatively large amounts of cholesterol. Foam cells also can derive from smooth muscle cells, as these cells can express scavenger receptors when appropriately activated.93,94 Formation of the fatty streak is thought to begin with adherence of circulating monocytes to activated endothelial cells at sites in the arterial system prone to atherosclerotic disease, such as at branch points in vessels. Multiple chemoattractant molecules have been identified in these nascent lesions, which recruit monocytes and induce their diapedesis into the subendothelial space where they further differentiate into macrophages. As noted above, however, more recent evidence indicates that microenvironment-supported macrophage proliferation in situ within the atherosclerotic lesion is likewise a key event in atherogenesis.84
The chemoattractant CCL2 (MCP-1) facilitates recruitment of monocytes to atherosclerotic lesions, as noted in studies of mouse models of atherosclerosis, such as apoE–/– or LDL receptor (LDLR–/–)–deficient mice fed a Western-style diet. When these mice are crossed to the model lacking CCL2, or its receptor CCR-2, lesion development decreases significantly.95,96,97
Macrophages and T cells were once thought to be the only inflammatory cells to significantly promote angiogenesis. More recent data showing that neutrophils are found at sites of plaque rupture or erosion and in thrombus from patients with acute coronary artery syndromes indicate that they also have an important role in atherothrombosis.18 Mast cells have been found in the adventitia of lesions and in areas of plaque hemorrhage; they have been implicated in macrophage apoptosis, increased vascular permeability, degradation of HDL and reduced cholesterol efflux.22 Neutrophils and mast cells are recruited to atherosclerotic lesions in response to CXC-chemokine receptor 2 (CXCR2) signals. The mobilization of neutrophils to atherosclerotic lesions is inhibited by CXC-chemokine receptor 4 (CXCR4) and its ligand CXC-chemokine ligand 12 (CXCRL12; also known as stromal-derived factor (SDF) 1. A subset of CD31+ T cells, so called Tang cells, was shown to promote endothelial repair and revascularization, and to be inversely correlated with age and cardiovascular disease (CVD) risk in patients undergoing coronary angiography.98
Macrophages control the amount of cholesterol loading by downregulating the native LDL receptor. Therefore, knowing how cholesterol is taken up into macrophages is important. Cell culture experiments revealed a “foam cell paradox,” in which macrophages engulf only modified lipids. Treatment of native LDL with copper or acetic anhydride (causing acetylation) led to increased LDL uptake through use of the scavenger receptor, leading to the formation of lipid-laden macrophages. These experiments led to the peroxidation theory of atherosclerosis,94 whereby LDL modification is an essential step in the development of foam cells. Although the precise mechanisms responsible for LDL oxidation remain unclear, enzymes including myeloperoxidase, inducible NO synthase, and NADPH oxidases are involved in the process.99,100 Of note, macrophages express each of these enzymes, which normally are used as antimicrobial reactive oxygen species essential for innate immunity.101 Thus, accumulation of cholesterol in the macrophage occurs via scavenger (not LDL) receptors of oxidized (and not native) LDL. Myeloperoxidase is an enzyme thought to cause lipid peroxidation in the intimal space and circulating levels are associated with adverse clinical outcomes in the setting of acute coronary syndromes and predictive of major adverse cardiovascular events.99
Conserved pattern recognition receptors expressed by macrophages include scavenger receptors A and B1 and CD36, all of which internalize oxidized LDL.102,103 Macrophages express various genes in response to oxidized LDL, including peroxisome proliferator-activated receptor-γ and adenosine triphosphate–binding cassette transporter A1, which profoundly influence macrophage-mediated inflammation and atherosclerotic activity.
Cell culture studies indicate that scavenger receptor A recognizes acetylated LDL but, unlike the LDL receptor, is not downregulated in response to increased cholesterol content and thus likely accounts for foam cell formation.104 However, no evidence indicates that acetyl LDL is generated in vivo, indicating other modifications of LDL, such as oxidation, may be required for foam cell formation.105,106 Another scavenger receptor presumed to be involved in the atherosclerotic process is CD36, a receptor that avidly binds oxidized LDL.
Circulating IgG and IgM antibodies against products of lipid peroxidation are present in the plasma of animals and humans.107 These antibodies closely correlate with measures of lipid peroxidation and with atherosclerotic progression and regression in murine models.108 Immunization of hypercholesterolemic rabbits and mice with products of oxidized LDL, such as malonyldialdehyde LDL or copper-oxidized LDL, inhibits the progression of atherosclerotic lesion formation.109,110,111,112 These experiments have been interpreted to indicate that an immunologic response to oxidized LDL components can alter the atherosclerotic process.
Leukocyte-derived 5-lipoxygenase also contributes to atherosclerosis susceptibility in mice.113 Animal studies indicate the importance of lipoxygenases in atherosclerosis as disruption of the 12,15-lipoxygenase gene diminishes atherosclerosis in apoE-deficient mice, and overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice.114,115 This enzyme is under study as a potential target to inhibit the atherosclerotic process.116
Gut Microbiome There are newer data indicating that intestinal microbes are involved in cardiometabolic diseases.117 Systemic inflammation is activated is the setting of chronic bacterial translocation (secondary to increased intestinal permeability) leading to macrophage influx into adipose tissue resulting in insulin resistance and nonalcoholic fatty liver disease. The increased inflammation may also be secondary to trimethylamine-N-oxide via influx of macrophages and cholesterol accumulation via upregulation of macrophage scavenger receptors and reduction in reverse cholesterol transport. Thus, gut microbiota may accelerate atherosclerosis risk.
Three potential factors lead to accumulation of LDL in the vascular wall: increased permeability of the endothelium, prolonged retention of lipoproteins in the intima, and slow removal of lipoproteins from the vessel wall.118 Rabbits fed a high-cholesterol diet develop aortic wall lesions at specific lesion-susceptible sites; however, endothelial permeability is not increased at those sites, indicating that LDL is selectively retained in these regions.119,120 Retention of LDL molecules likely results from their adherence to proteoglycans in the vessel wall.121 LDL genetically engineered to not bind to proteoglycans is hypothesized to be less atherogenic than native LDL.20
Oxidized LDL and its products, oxidized phospholipids and oxysterols, have other properties that make them potentially proatherogenic.122 These properties include proinflammatory characteristics, such as chemotactic signaling for monocytes, smooth muscle cells, and T lymphocytes (but not for B lymphocytes or neutrophils, neither of which is found in lesions) and increased expression of VCAM-1 on, and stimulation of CCL2 release from, endothelial cells.123 Oxidized LDL also may contribute to instability of the atherosclerotic plaque via induction of type 1 metalloproteinase expression and increase in TF activity.124 For oxidized LDL to be a ligand for the scavenger receptor, extensive degradation of the polyunsaturated fatty acid in the sn-2 position of phospholipids by oxidation is essential.
To test the oxidized LDL hypothesis, several clinical studies have been conducted using antioxidant vitamins, most commonly vitamin E; however, most of the completed studies gave negative results.125,126 At the present time, treatment with vitamin E does not appear to be beneficial in preventing cardiovascular events.
A low level of HDL cholesterol is a strong predictor of adverse cardiovascular events, presumably because the low level is associated with insufficient reverse cholesterol transport. Animal studies using liver-directed gene transfer of human apoA–apoI resulted in significant promotion of reverse cholesterol transport and regression of preexisting atherosclerotic lesions in LDL receptor-deficient mice.127,128 The HDL level may not be as important as amount of reverse cholesterol transport. For example, the capacity of HDL to accept cholesterol from macrophages is predictive of atherosclerotic burden.129 However, HDL has additional antiatherogenic properties that may confer protection against atherosclerosis.130 For example, HDL is protective against oxidation of LDL, at least in part because of paraoxonase, an enzyme physically associated with HDL that degrades organophosphates.131 Paraoxonase polymorphisms are associated with increased risk of CVD, also indicating that oxidized LDL is an important factor in atherosclerotic development.132
Research studies currently are evaluating novel ways to increase HDL levels or to use apoA–apoI variants and mimetics that hopefully will cause regression of atherosclerosis. So far, initial clinical studies were not successful. Cholesteryl ester transfer protein promotes the transfer of cholesteryl esters from antiatherogenic HDLs to proatherogenic apoB-containing lipoproteins, including very-low-density lipoproteins (VLDLs), VLDL remnants, intermediate-density lipoproteins, and LDLs. A deficiency of this molecule results in increased HDL levels and decreased LDL levels, a lipid profile that is antiatherogenic. A large clinical study in humans showed that inhibition of the transfer protein with torcetrapib increased HDL levels but was associated with increased mortality and hypertension.133 It is supposed that the increase in mortality was a result of an off-target effect of the drug increasing blood pressure and not because of cholesterol ester transfer protein inhibition. Clinical trials evaluating the effect of other inhibitors of cholesteryl ester transfer protein on atherosclerosis and cardiovascular events have not shown clinical efficacy thus far. Along similar lines, delivery of a mutant form of apoA1 (apoA1 Milano) resulted in regression of plaque size as measured by intravascular ultrasound in a small phase II clinical trial.134 However, studies evaluating the effect of apoA1 mimetics on atherosclerosis also are have not shown compelling data regarding reducing plaque size or cardiovascular events.135
Studies indicate that human atherosclerotic lesions express the immune mediator CD40 and its soluble ligand, sCD40L. Increasing evidence indicates that the CD40–sCD40L signaling pathway plays a central role in several inflammatory processes, including atherosclerosis and graft rejection following transplantation.136 Interruption of CD40 signaling in hyperlipidemic mice reduces the size of aortic atherosclerotic lesions and their lipid, macrophage, and T-lymphocyte content.137 Atorvastatin, lovastatin, pravastatin, and simvastatin reduce IFN-γ–induced CD40 expression in a dose-dependent manner. Activation of atheroma-associated cells with human recombinant sCD40L is reduced when cells are treated with statins. In addition, retrospective ex vivo immunostaining of human carotid atherosclerotic lesions of patients treated with simvastatin for more than 3 months revealed less CD40 expression and atheroma-associated cells compared with patients who were not treated with the drug. A reduction in sCD40L is associated with pravastatin or cerivastatin therapy.138 These findings support the notion that statins are antiinflammatory, in addition to their cholesterol-lowering effects.
Transforming growth factor (TGF)-β is a cytokine secreted by macrophages, smooth muscle cells, and the Th3 subset of Th cells that has multiple regulatory functions. TGF-β is speculated to contribute to plaque stabilization because it stimulates collagen synthesis and is fibrogenic. One study found that inhibition of TGF-β signaling by neutralizing antibodies led to a larger plaque size with an unstable phenotype.139 Further studies are needed to clarify the role of TGF-β in atherosclerotic plaque initiation and growth.
ABO Blood Type and Cardiovascular Risk The ABO blood group is determined from presence of A and B antigens on the surface of the red blood cells and is thought to confer cardiovascular risk.140 These antigens are also expressed on the surface of platelets and the endothelium and consist of terminal carbohydrate molecules which are synthesized by the sequential action of the ABO glycosyltransferases. Genetic studies showed that carriers of single nucleotide polymorphisms (SNPs) that mark non-O blood group types have higher levels of plasma VWF when compared to O individuals. Epidemiologic and genetic studies show that the non-O blood group is associated with adverse cardiovascular events. One study demonstrated that the SNP rs514659 was associated with coronary artery diseases (CADs) when complicated by MI but not with CAD without MI, suggesting that the primary relationship of ABO to clinical CAD is through modulation of coronary thrombosis or plaque rupture in patients with established coronary atherosclerosis rather than through primary promotion of atherosclerosis per se.
Several infectious agents have been implicated as pathogens in atherosclerosis.141 A well-studied infectious pathogen is C. pneumoniae. Animals infected with this agent develop atherosclerosis, and patients with CVD have higher titers of antibodies against this pathogen. Viruses, such as herpes simplex and cytomegalovirus, also are implicated in human atherosclerotic lesion formation. Gingivitis as a consequence of poor dental hygiene or smoking may lead to cellular immune activation and provoke atherosclerosis by cytokines or antibodies.142 Endogenous proteins, such as heat shock proteins, also are implicated in atherosclerosis. One study showed that progression of carotid disease correlated with antibodies against heat shock proteins 65 and 60.143
The relationship between the immune system and atherosclerosis is complex, as evident from an animal study that showed that splenectomy of cholesterol-fed apoE–/– mice led to significantly increased atherosclerosis.144 This proatherogenic effect was rescued by transfer of either purified B cells or T cells from the spleens of atherosclerotic apoE–/– donors. A long-term study of soldiers who underwent splenectomy after trauma found the soldiers had a twofold increased incidence of CAD, providing evidence that the spleen has antiatherogenic activity.145 Further studies are needed to determine if splenectomy significantly impacts the atherosclerotic process, and if so, by what mechanism.
Atherosclerotic disease is a complex human trait involving multiple genes and environmental factors. Through the study of linkage analysis of families and sibling pairs as well as candidate genes and genome-wide association studies, the genetic predisposition to MI is starting to be understood.146 The clinical importance of this knowledge is the potential identification of markers of disease for risk prediction and potential intervention to lower the risk of atherosclerotic-based cardiovascular events.
The use of genome-wide linkage analyses of families or sib-pairs has identified chromosomal loci linked to or genetic variations in the arachidonic 5-lipoxygenase-activating protein gene (ALOX5AP)147 and leukotriene A4 hydrolase gene (LTA4H).148 The genes are both involved in inflammation-related pathway of leukotriene B4 production. Interestingly, a small molecule inhibitor of ALOX5AP was shown to reduce leukotriene production and plasma levels of C-reactive protein.
Several association studies of unrelated individuals have identified genetic variations that confer susceptibility to atherosclerotic disease and cardiovascular events. Studies using genome-wide linkage analysis identified four SNPs on chromosome 9p21.3 that were associated with MI in white cohorts.146,149 Other genetic polymorphisms also contribute to increased risk of CVD through a variety of mechanisms.150
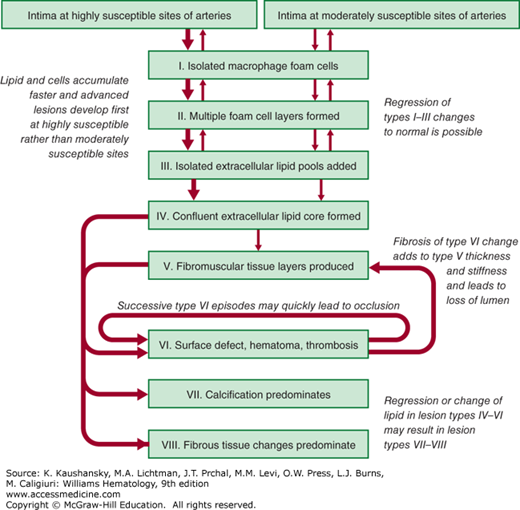
The American Heart Association classification of atherosclerotic plaques into types I through VIII is based on lesion composition and structure (Fig. 134–5).23 Types I through III atherosclerotic plaques have foam cells organized in a fatty streak, ranging from those not visible on close examination (type I) to those that are apparent on examination (type III). Types I through III lesions are small and clinically silent, whereas types IV through VI lesions may obstruct the lumen and produce a clinical event. Type IV lesions contain a confluent pool of lipid and in most patients do not cause anginal symptoms because of the ability of the artery to remodel outward. Type V lesions contain a fibromuscular cap resulting from replacement of tissue disrupted by accumulated lipid and hematoma or organized thrombotic deposits. Type VI lesions involve thrombosis that may be either mural or obstructive. Of note, a type IV lesion may develop type VI changes without ever passing through a type V change and accumulating significant fibrous tissue. Plaques that are complex and primarily composed of calcium are type VII lesions or, if fibrous tissue predominates, are type VIII lesions.
Figure 134–5.
Flow diagram in center column indicates pathways in evolution and progression of human atherosclerotic lesions. Roman numerals indicate histologically characteristic types of lesions. The direction of arrows indicates sequence in which characteristic morphologies may change. From type I to type IV, changes in lesion morphology occur primarily because of increasing accumulation of lipid. The loop between types V and VI illustrates how lesions increase in surfaces. Thrombotic deposits may develop repeatedly over varied time spans in the same location and may be the principal mechanism for gradual occlusion of medium-sized arteries.
The pathologic mechanisms responsible for converting chronic coronary atherosclerosis to an acute coronary event result, in part, from plaque disruption, a term that was synonymously used with plaque rupture.151,152 The term vulnerable plaque was used by Muller and colleagues153,154 to describe rupture-prone plaques as the underlying cause of most clinical coronary events. The current definition for “vulnerable plaque” includes all thrombosis-prone plaques and those with a high probability of undergoing rapid progression, thus becoming culprit plaques (Fig. 134–6).155
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


